Temporal Transcriptome Change of Oncomelania
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Resettlement Plan for Medium City Traffic Construction Project of Anhui Province
RP825 V1 World Bank Financed Project Public Disclosure Authorized Resettlement Plan for Medium City Traffic Construction Project of Anhui Province Public Disclosure Authorized (Summary Report) Public Disclosure Authorized Public Disclosure Authorized July 2009 Final Report of Resettlement Action Plan for World Bank Financed Medium City Traffic Construction Project of Anhui Province Terms and Definitions I. Displaced persons 1. Displaced persons (DPs) may be classified in one of the following three groups by eligibility for compensation: A. those who have formal legal rights to land (including customary and traditional rights recognized under the laws of the country); B. those who do not have formal legal rights to land at the time the census begins but have a claim to such land or assets—provided that such claims are recognized under the laws of the country or become recognized through a process identified in the resettlement plan; and C. those who have no recognizable legal right or claim to the land they are occupying. 2. Persons covered under para. 2(A) and (B) are provided compensation for the land they lose, and other assistance. Persons covered under para. 2(C) are provided resettlement assistance in lieu of compensation for the land they occupy, and other assistance, as necessary, to achieve the objectives set out in this policy, if they occupy the project area prior to a cut-off date1 established by the borrower and acceptable to the Bank. Persons who encroach on the area after the cut-off date are not entitled to compensation or any other form of resettlement assistance. All persons included in para. -

中国输美木制工艺品注册登记企业名单registered Producing
中国输美木制工艺品注册登记企业名单 Registered Producing Industries of Wooden Handicrafts of China Exported to the U.S. 注册登记编号 序号 所在省份 所在城市 企业名称 企业地址 Registered Number Province City Company Name Company Address Number 天津市北辰区双口镇双河村30号 天津津长工艺品加工厂 天津 天津 NO.30, SHUANGHE VILLAGE, 1 TIANJIN JINCHANG ARTS & 1206ZMC0087 TIANJIN TIANJIN SHUANGKOU TOWN, BEICHEN CRAFTS FACTORY DISTRICT, TIANJIN CITY, CHINA 天津市静海县利邦工艺制品厂 天津市静海区西翟庄镇西翟庄 天津 天津 2 TIANJIN JINGHAI LEBANG ARTS XIZHAI ZHUANG, JINGHAI COUNTRY 1204ZMC0004 TIANJIN TIANJIN CRAFTS CO.,LTD TIANJIN 天津市宁河县板桥镇田庄坨村外东侧 天津鹏久苇草制品有限公司 天津 天津 THE EAST SIDE OF TIANZHUANGTUO 3 TIANJIN PENGJIU REED PRODUCTS 1200ZMC0012 TIANJIN TIANJIN VILLAGE, BANQIAO TOWN, NINGHE CO.,LTD. COUNTY, TIANJIN, CHINA 河北百年巧匠文化传播股份有限公司 石家庄桥西区新石北路399号 河北 石家庄 4 HEBEI BAINIANQIAOJIANG NO.399, XINSHI NORTH ROAD, QIAOXI 1300ZMC9003 HEBEI SHIJIAZHUANG CULTURE COMMUNICATION INC. DISTRICT, SHIJIAZHUANG CITY 行唐县森旺工贸有限公司 河北省石家庄行唐县口头镇 河北 石家庄 5 XINGTANGXIAN SENWANG KOUTOU TOWN, XINGTANG COUNTY, 1300ZMC0003 HEBEI SHIJIAZHUANG INDUSTRY AND TRADING CO.,LTD SHIJIAZHUANG, HEBEI CHINA 唐山市燕南制锹有限公司 河北省滦南县城东 河北 唐山 6 TANGSHAN YANNAN EAST OF LUANNAN COUNTY, HEBEI, 1302ZMC0002 HEBEI TANGSHAN SHOVEL-MAKING CO.,LTD CHINA 唐山天坤金属工具制造有限公司 河北省唐山市滦南县唐乐公路南侧 河北 唐山 7 TANGSHAN TIANKUN METAL SOUTH OF TANGLE ROAD, LUANNAN 1302ZMC0008 HEBEI TANGSHAN TOOLS MAKING CO.,LTD COUNTY, HEBEI, CHINA 唐山市长智农工具设计制造有限公司 TANGSHAN CHANGZHI 河北省滦南县城东杜土村南 河北 唐山 8 AGRICULTURAL TOOLS SOUTH OF DUTU TOWN, LUANNAN 1302ZMC0031 HEBEI TANGSHAN DESIGNING AND COUNTY, HEBEI, CHINA MANUFACTURING CO.,LTD. 唐山腾飞五金工具制造有限公司 河北省滦南县宋道口镇 河北 唐山 9 TANGSHAN TENGFEI HARDWARE SONG DAO KOU TOWN, LUANNAN 1302ZMC0009 HEBEI TANGSHAN TOOLS MANUFACTURE CO.,LTD. COUNTY, HEBEI CHINA 河北省滦南县宋道口镇杜土村南 唐山舒适五金工具制造有限公司 河北 唐山 SOUTH OF DUTU, SONG DAO KOU 10 TANGSHAN SHUSHI HARDWARE 1302ZMC0040 HEBEI TANGSHAN TOWN, LUANNAN COUNTY, HEBEI TOOLS MANUFACTURE CO,LTD CHINA 唐山腾骥锻轧农具制造有限公司 河北省滦南县宋道口镇 河北 唐山 TANGSHAN TENGJI FORGED 11 SONG DAO KOU TOWN, LUANNAN 1302ZMC0011 HEBEI TANGSHAN AGRICULTURE IMPLEMENTS COUNTY, HEBEI CHINA MANUFACTURING CO.,LTD. -

In Bahia, Brazil
Volume 52(40):515‑524, 2012 A NEW GENUS AND SPECIES OF CAVERNICOLOUS POMATIOPSIDAE (MOLLUSCA, CAENOGASTROPODA) IN BAHIA, BRAZIL 1 LUIZ RICARDO L. SIMONE ABSTRACT Spiripockia punctata is a new genus and species of Pomatiopsidae found in a cave from Serra Ramalho, SW Bahia, Brazil. The taxon is troglobiont (restricted to subterranean realm), and is characterized by the shell weakly elongated, fragile, translucent, normally sculptured by pus‑ tules with periostracum hair on tip of pustules; peristome highly expanded; umbilicus opened; radular rachidian with 6 apical and 3 pairs of lateral cusps; osphradium short, arched; gill filaments with rounded tip; prostate flattened, with vas deferens inserting subterminally; penis duct narrow and weakly sinuous; pallial oviduct simple anteriorly, possessing convoluted by‑ pass connecting base of bulged portion of transition between visceral and pallial oviducts with base of seminal receptacle; spermathecal duct complete, originated from albumen gland. The description of this endemic species may raise protective environmental actions to that cave and to the Serra Ramalho Karst area. Key-Words: Pomatiopsidae; Spiripockia punctata gen. nov. et sp. nov.; Brazil; Cave; Tro- globiont; Anatomy. INTRODUCTION An enigmatic tiny gastropod has been collected in caves from the Serra Ramalho Kars area, southwestern The family Pomatiopsidae is represented in the Bahia state, Brazil. It has a pretty, fragile, translucent Brazilian region by only two species of the genus Id‑ shell in such preliminary gross anatomy, which already iopyrgus Pilsbry, 1911 (Simone, 2006: 94). However, reveals troglobiont adaptations, i.e., depigmentation, the taxon is much richer in remaining mainland ar- lack of eyes and small size. The sample has been brought eas, with both freshwater and semi-terrestrial habits by Maria Elina Bichuette, who is specialized in subter- (Ponder & Keyzer, 1998; Kameda & Kato, 2011). -
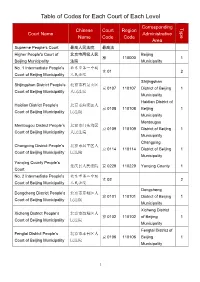
Table of Codes for Each Court of Each Level
Table of Codes for Each Court of Each Level Corresponding Type Chinese Court Region Court Name Administrative Name Code Code Area Supreme People’s Court 最高人民法院 最高法 Higher People's Court of 北京市高级人民 Beijing 京 110000 1 Beijing Municipality 法院 Municipality No. 1 Intermediate People's 北京市第一中级 京 01 2 Court of Beijing Municipality 人民法院 Shijingshan Shijingshan District People’s 北京市石景山区 京 0107 110107 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Haidian District of Haidian District People’s 北京市海淀区人 京 0108 110108 Beijing 1 Court of Beijing Municipality 民法院 Municipality Mentougou Mentougou District People’s 北京市门头沟区 京 0109 110109 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Changping Changping District People’s 北京市昌平区人 京 0114 110114 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Yanqing County People’s 延庆县人民法院 京 0229 110229 Yanqing County 1 Court No. 2 Intermediate People's 北京市第二中级 京 02 2 Court of Beijing Municipality 人民法院 Dongcheng Dongcheng District People’s 北京市东城区人 京 0101 110101 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Xicheng District Xicheng District People’s 北京市西城区人 京 0102 110102 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Fengtai District of Fengtai District People’s 北京市丰台区人 京 0106 110106 Beijing 1 Court of Beijing Municipality 民法院 Municipality 1 Fangshan District Fangshan District People’s 北京市房山区人 京 0111 110111 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Daxing District of Daxing District People’s 北京市大兴区人 京 0115 -

Transcriptome Sequencing and Differential Gene Expression
www.nature.com/scientificreports OPEN Transcriptome sequencing and diferential gene expression analysis of the schistosome- Received: 26 May 2017 Accepted: 7 November 2017 transmitting snail Oncomelania Published: xx xx xxxx hupensis inhabiting hilly and marshland regions Jin-Song Zhao1, An-Yun Wang2, Hua-Bin Zhao3 & Yan-Hong Chen3 The freshwater snail Oncomelania hupensis is the unique intermediate host of the blood fuke Schistosoma japonicum, which is the major cause of schistosomiasis. The snail inhabits two contrasting environments: the hilly and marshland regions. The hilly snails are smaller in size and have the typical smooth shell, whereas the marshland snails are larger and possess the ribbed shell. To reveal the diferences in gene expression between the hilly and marshland snails, a total of six snails, three per environment, were individually examined by RNA sequencing technology. All paired-end reads were assembled into contigs from which 34,760 unigenes were predicted. Based on single nucleotide polymorphisms, principal component analysis and neighbor-joining clustering revealed two distinct clusters of hilly and marshland snails. Analysis of expression changes between environments showed that upregulated genes relating to immunity and development were enriched in hilly snails, while those associated with reproduction were over-represented in marshland snails. Eight diferentially expressed genes between the two types of snails were validated by qRT-PCR. Our study identifed candidate genes that could be targets for future functional studies, and provided a link between expression profling and ecological adaptation of the snail that may have implications for schistosomiasis control. Te blood fuke Schistosoma japonicum (Platyhelminth: Trematoda) occurs in China and, to a lesser extent, in the Philippines and parts of Indonesia, and human infection by the blood fuke causes a major public health problem especially in lake and marshland regions1,2. -

Genetic Comparison of Oncomelania Hupensis Quadrasi (Möllendorf
SGeneticCIENCE D ComparisonILIMAN (JULY -DofECEMBER Oncomelania 2017) 29:2, hupensis 32-50 quadrasi Genetic Comparison of Oncomelania hupensis quadrasi (Möllendorf, 1895) (Gastropoda: Pomatiopsidae), the Intermediate Host of Schistosoma japonicum in the Philippines, Based on 16S Ribosomal RNA Sequence James Christopher C. Chua* Ian Kim B. Tabios Pebbles Grayle P. Tamayo Lydia R. Leonardo University of the Philippines Manila Ian Kendrich C. Fontanilla Raffy Jay C. Fornillos University of the Philippines Diliman Emmanuel Ryan C. De Chavez University of the Philippines Los Baños Takeshi Agatsuma Kochi Medical School Mihoko Kikuchi Nagasaki University Naoko Kato-Hayashi Yuichi Chigusa Dokkyo Medical University ABSTRACT Schistosomiasis japonica is a water-borne trematode infection transmitted by different subspecies of Oncomelania hupensis. As parasites may either co-evolve or locally adapt with their hosts, snail diversity, as revealed by morphometric and genetic studies, may reflect parasite diversity and elucidate snail susceptibility and transmission patterns. This study aimed to compare isolates of O. h. quadrasi based on a 342-bp fragment of _______________ *Corresponding Author ISSN 0115-7809 Print / ISSN 2012-0818 Online 32 J.C. C. Chua et al. the 16S ribosomal RNA gene. O. h. quadrasi isolates were collected from nine provinces known to have S. japonicum in the Philippines, namely Cagayan Valley, Bohol, Negros Occidental, Leyte, Davao, Davao del Sur, Mindoro Oriental, Northern Samar, and Sorsogon. O. h. hupensis and O. h. nosophora isolates were also collected from China and Japan, respectively. The 16S ribosomal RNA gene of each specimen was amplified and sequenced. Phylogenetic and network analyses based on the 221 16S rRNA gene sequences revealed that O. -

World Bank Document
RP174 V. 4 July 31, 2003 10/10/2002 Public Disclosure Authorized Appraisal Report for Relief of Poverty at Affected Areas by Public Disclosure Authorized Anhui Highway Project II Public Disclosure Authorized China Cross-cultural Center, Zhongshan University July, 31, 2002 Public Disclosure Authorized Guangzhou, China FiLE Cji.e.sy TABLE OF CONTENTS Chapter 1 General 1. Introduction to Poverty Appraisal on Affected Areas 2. Poverty and Appraisal 3. Objective and Methodology of Poverty Appraisal Chapter 1 Background of Affected Areas 1. Geographic Location and Human Cultural Characteristics of Affected Areas 2. Advantages of Natural and Human Resource at Affected Areas 3. Direct/Indirect Beneficiaries at Affected Areas 4. Current Traffic Conditions at Affected Areas Chapter 2 Background of Affected Areas 1. Distribution of Poor Population at Affected Areas 2. Social and Economic Developments of Affected Areas 3. Analysis of Poverty Causes at Affected Areas Chapter 3 Analysis of Poverty Situations and Poverty Causes at Affected Areas Chapter 4 Antipoverty Measures Already Executed at Affected Areas and Social/Economic Benefits Chapter 5 Possible Antipoverty Effects from Project Execution Chapter 6 Benefited Groups' Attitudes towards Project Execution at Affected Areas Chapter 7 Opinions and Suggestions Chapter 1 General 1. Introduction to Poverty Appraisal on Affected Areas This appraisal is a topical appraisal based on the "social appraisal". Entrusted by the World Bank Financed Project Execution Office (PEO) under the Anhui Provincial Communications Department (APCD), the CCCC at Zhongshan University conducted an independent social appraisal on the proposed "Anhui Provincial Highway II Project & Local Road Improvement Program (AHPII& RRIP)", including the Road Safety Project (RSP) during May 16-26, 2002. -

Assessment of the Transmission Risk of Schistosomiasis After Flooding — North Poyang Lake, Jiangxi Province, China, 2020
China CDC Weekly Preplanned Studies Assessment of the Transmission Risk of Schistosomiasis after Flooding — North Poyang Lake, Jiangxi Province, China, 2020 Shan Lv1,#; Fan Yang1; Zhiqiang Qin1; Chunli Cao1; Jing Xu1; Shizhu Li1; Xiao-nong Zhou1 risk of schistosomiasis transmission (1). A risk Summary assessment in Jiangxi was conducted for schistosomiasis What is already known about this topic? transmission and provided interventions after flooding. Over 90% of Oncomelania snails, the only intermediate Human stool samples, Oncomelania snails, and animal host of Schistosoma japonicum, are distributed in the feces released to the environment were collected in two middle and low reaches of Yangtze River. Flooding can counties, i.e., Lushan and Lianxi. Although no infected extend the distribution of Oncomelania snails and humans were found in the survey, infections in snails hence accelerate the transmission of schistosomiasis. and animal feces were discovered from four field sites What is added by this report? in Lushan. Our results indicated the intensive Although the dispersal of Oncomelania snails was surveillance should be implemented post-flooding. negligible in north Poyang Lake after flooding in 2020, Two neighboring counties, i.e., Lushan and Lianxi, 2 samples of cattle feces with Schistosoma egg and 2 located in the northwest bank of Poyang Lake leading infected snails samples were indeed found. All four risk to Yangtze River were selected. The evidence showed sites were distributed in Lushan County. Cattle feces that the density of Oncomelania snails increased were observed in the six out of seven field sites in significantly and exceeded that in the south lake Lushan County. -

Phylogenetic Relationships of Snails of the Genera Oncomelania and Tricula Inferred from the Mitochondrial 12S Rrna Gene
Jpn. J. Trop. Med。 Hyg., Vol.31, No.1,2003, pp.5-10 5 Phylogenetic relationships of snails of the genera Oncomelania and Tricula inferred from the mitochondrial 12S rRNA gene MUNEHIRO OKAMOTO', CHIN-TSON L02, WILFRED U. TIU3, DONGCHUAN QUI4, PINARDI HADIDJAJA5,SUCHART UPATHAM6, HIROMU SUGIYAMA7, TAKAHIROTAGUCHI8, HIROHISA HIRAI9,YASUHIDE SAITOW10, SHIGEHISAHABE", MASANORI KAWANAKA7,MIZUKI HIRATA12AND TAKESHIAGATSUMA13* Accepted 28, February, 2002 Abstract The Schistosoma japonicum group and S. sinensium utilize intermediate snail hosts belonging to the genera Oncomelania and Tricula (Gastropoda: Pomatiopsidae). In the present study, partial sequences of the mitochon- drial 12S rRNA gene from 7 subspecies of 0. hupensis, two species of Tricula (T bollingi and T humida) and 0. minima were examined to infer a phylogeny for these. Nucleotide differences among subspecies of 0. hupensis were less than 6.5% and among species from different genera, 10-12%. The phylogenetic tree obtained in this study indicates that 0. hupensis subspecies fell into four distinct clades ; that is, 0. h. quadrasi from the Philip- pines, 0. h. lindoensis from Indonesia, 0. h. hupensis from Yunnan, China and the remaining 5 subspecies (0. h. hupensis from other parts of China, 0. h. robertsoni from China, 0. h. formosana from Taiwan, 0. h. chiui from Taiwan and 0. h. nosophora from Japan). The phylogenetic tree also showed that 0. minima was placed as sister to all of the subspecies of 0. hupensis. Possible evolutionary relationships among the snail hosts were discussed. Key Words: Oncomelania, Tricula, mitochondrial DNA, 12S rRNA gene, phylogenetic tree class Pulmonata, while pomatiopsids belong to the subclass INTRODUCTION Caenogastropoda. -

THE STATUS and DISTRIBUTION of Freshwater Biodiversity in Madagascar and the Indian Ocean Islands Hotspot
THE THE STATUs aNd dISTRIBUtION OF STAT U Freshwater biodIversIty in MadagasCar s a N aNd the INdIaN OCeaN IslaNds hOtspOt d d I STR Edited by Laura Máiz-Tomé, Catherine Sayer and William Darwall IUCN Freshwater Biodiversity Unit, Global Species Programme IBU t ION OF F OF ION RESHWATER N ds a BIO I N d I ar ar VERS d C N I TY IN IN sla Madagas I N C ar a ar N ea d the I the d d the I the d C N N d Madagas a O I a N O C ea N I sla N IUCN h ds Rue Mauverney 28 CH-1196 Gland O Switzerland tsp Tel: + 41 22 999 0000 Fax: + 41 22 999 0015 O www.iucn.org/redlist t the IUCN red list of threatened speciestM www.iucnredlist.org THE STATUS AND DISTRIBUTION OF freshwater biodiversity in Madagascar and the Indian Ocean islands hotspot Edited by Laura Máiz-Tomé, Catherine Sayer and William Darwall IUCN Freshwater Biodiversity Unit, Global Species Programme The designation of geographical entities in this book, and the presentation of the material, do not imply the expression of any opinion whatsoever on the part of IUCN concerning the legal status of any country, territory, or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. The views expressed in this publication do not necessarily reflect those of IUCN, or other participating organisations. This publication has been made possible by funding from The Critical Ecosystem Partnership Fund. Published by: IUCN Cambridge, UK in collaboration with IUCN Gland, Switzerland Copyright: © 2018 IUCN, International Union for Conservation of Nature and Natural Resources Reproduction of this publication for educational or other non-commercial purposes is authorised without prior written permission from the copyright holder provided the source is fully acknowledged. -

Change Pattern and Driving Mechanism of Construction Land In
EARTH SCIENCES RESEARCH JOURNAL LAND USE Earth Sci. Res. J. Vol. 24, No. 2 (June, 2020): 215-223 Change pattern and driving mechanism of construction land in China’s undertaking industrial transfer demonstration area: Taking the Wanjiang City Belt along the Yangtze River as an Example Yuhong Cao1*, Meiyun Liu1, Yuandan Cao1, Chen Chen1, Dapeng Zhang2 1School of Environmental Science and Engineering, Anhui Normal University, Wuhu 241002, China 2School of Geography and Tourism, Anhui Normal University, Wuhu 241002, China * Corresponding author: [email protected] ABSTRACT Keywords: expansion intensity; land use; hotspot The construction land includes urban land, rural residential areas, and other construction lands. The Wanjiang City Belt analysis; the Wanjiang City Belt along the along the Yangtze River is a vital demonstration area for undertaking industrial transfer in China. With the accumulation Yangtze River. of factors relative to economic development, the construction land has increased sharply, and the regional ecological security pattern is facing new challenges. After collecting the image interpretation data of multi-period land use of the Wanjiang City Belt, this work studies the characteristics of construction land change patterns since 1995. The driving mechanism was also analyzed based on the GIS platform, land use transfer matrix, expansion intensity index, hotspot analysis, and mathematical statistics. The results showed that: (1) From 1995 to 2015, the urban land and other construction lands in the Wanjiang City Belt have increased, but the rural residential areas decreased in 2010- 2015. The three types of lands had the most significant changes in 2005-2010, and the other construction land was particularly prominent. -

Species : Oncomelania Hupensis Quadrasi
Draft risk assessment report addressing Terms of Reference Species : Oncomelania hupensis quadrasi 1. Taxonomy of the species Phylum Mollusca Class Gastropoda Family Pomatiopsidae Oncomelania hupensis quadrasi also known as Oncomelania quadrasi is a subspecies of Oncomelania hupensis – wild type strains only. Another subspecies (Oncomelania hupensis hupensis) is already on the Live Import List. 2. Status of species under CITES This species is prevalent in large numbers in several regions of the Philippines [1-4]. The species is not listed in CITES. The snails to be imported are derived from laboratory stocks maintained in the Philippines and/or the USA. The laboratory strain has been obtained from wild populations in the Philippines and has not been genetically modified. Occasionally, it may be necessary to source infected snails from wild populations to ensure the lab stocks do not become less pathogenic than endemic isolates. 3. Ecology of the species Oncomelania quadrasi is a tropical, freshwater snail that is operculated, amphibious and dioecious [1,2,4]. It feeds on green algae, diatoms and decaying vegetative matter. The snail lives in wet environments such as flood plain forests, swamps and sluggish streams, ones usually clogged with vegetation [1,2,4]. The species is susceptible to desiccation in the absence of moisture for prolonged periods [1,2,4]. Life Span: The snail can live for about 4-6 months in the wild, though it can live substantially longer in laboratory conditions. Those snails used to maintain the Schistosoma japonicum parasite life cycle in the laboratory will be crushed to harvest the parasite after 3 months post infection [1].