Abstract of Thesis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Circular of the Bureau of Standards No. 539 Volume 10: Standard X-Ray
:ationa.u d H.W. BIS <T be Libra.ry, Reisrence book not to 1965 JVPR 1 6 from ibe lib s ary. taken NBS C | RCULAR 539 VOLUME 10 Standard X-ray Diffraction Powder Patterns UNITED STATES DEPARTMENT OF COMMERCE NATIONAL BUREAU OF STANDARDS THE NATIONAL BUREAU OF STANDARDS Functions and Activities The Functions of the National Bureau of Standards are set forth in the Act of Congress, March 3, 1901, as amended by Congress in Public Law 619, 1950. These include the development and maintenance of the national standards of measurement and the provision of means and methods for making measurements consistent with these standards; the determination of physical constants and properties of materials; the development of methods and instruments for testing materials, devices, and structures; advisory services to government agencies on scientific and technical problems; in- vention and development of devices to serve special needs of the Government; and the development of standard practices, codes, and specifications. The work includes basic and applied research, development, engineering, instrumentation, testing, evaluation, calibration services, and various consultation and information services. Research projects are also performed for other government agencies when the work relates to and supplements the basic program of the Bureau or when the Bureau’s unique competence is required. The scope of activities is suggested by the listing of divisions and sections on the inside of the back cover. Publications The results of the Bureau’s work take the form of either actual equipment and devices or pub- lished papers. These papers appear either in the Bureau’s own series of publications or in the journals of professional and scientific societies. -

WO 2015/121485 Al 20 August 2015 (20.08.2015) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2015/121485 Al 20 August 2015 (20.08.2015) P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every B01J 23/18 (2006.01) B01J 35/02 (2006.01) kind of national protection available): AE, AG, AL, AM, B01J 23/22 (2006.01) C07C 15/24 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, B01J 27/198 (2006.01) B01J 37/00 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, B01J 35/00 (2006.01) DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, (21) International Application Number: KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, PCT/EP2015/053270 MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (22) International Filing Date: PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, 17 February 2015 (17.02.2015) SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every (26) Publication Language: English kind of regional protection available): ARIPO (BW, GH, (30) Priority Data: GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, 14155332. -

CLARC Excerpt
Washington State Department of Ecology - CLARC Air Table (Methods B and C) - February 2021 February 2021 S S CPFi S CPFo S Air Air RfC o RfDi o Inhalation RfDo o Oral o Air Air Method C Method C Inhalation u Inhalation IUR u Cancer Oral u Cancer u Method B Method B Noncancer Cancer Reference Reference Inhalation Potency Reference Potency Noncancer Cancer (Eq. 750-1 (Eq. 750-2 Chemical Data Links to r r r r Concentration c Dose Unit Risk c Factor Dose c Factor c (Eq. 750-1) (Eq. 750-2) adjusted) adjusted) 3 3 -1 CAS No. Group Chemical Name Important Notes (mg/m ) e (mg/kg-day) (µg/m ) e (kg-day/mg) (mg/kg-day) e (kg-day/mg) e (µg/m³) (µg/m³) (µg/m³) (µg/m³) 83-32-9 PAHs acenaphthene 6.00E-02 I 30560-19-1 Pesticides acephate 1.20E-03 O 75-07-0 VOCs acetaldehyde 9.00E-03 I 2.57E-03 2.20E-06 I 7.70E-03 4.10E+00 1.10E+00 9.00E+00 1.10E+01 34256-82-1 Pesticides acetochlor 2.00E-02 I 67-64-1 VOCs acetone 3.10E+01 A 8.86E+00 9.00E-01 I 1.40E+04 3.10E+04 75-86-5 VOCs acetone cyanohydrin 2.00E-03 X 5.71E-04 9.10E-01 2.00E+00 75-05-8 VOCs acetonitrile 6.00E-02 I 1.71E-02 2.70E+01 6.00E+01 98-86-2 SVOCs acetophenone 1.00E-01 I 62476-59-9 Herbicides acifluorfen, sodium 1.30E-02 I 107-02-8 VOCs acrolein 2.00E-05 I 5.71E-06 5.00E-04 I 9.10E-03 2.00E-02 79-06-1 VOCs acrylamide 6.00E-03 I 1.71E-03 1.00E-04 I-M 3.50E-01 2.00E-03 I 5.00E-01 I-M 2.70E+00 6.60E-03 6.00E+00 2.50E-01 79-10-7 VOCs acrylic acid 1.00E-03 I 2.86E-04 5.00E-01 I 4.60E-01 1.00E+00 107-13-1 VOCs acrylonitrile 2.00E-03 I 5.71E-04 6.80E-05 I 2.38E-01 4.00E-02 A 5.40E-01 I 9.10E-01 3.70E-02 -
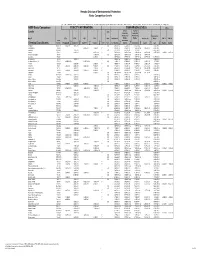
Basic Comparison Levels
Nevada Division of Environmental Protection Basic Comparison Levels Key: I=IRIS; P= PPRTV; N=NCEA; H=HEAST; A=ATSDR; O=Other Documents; CA=CalEPA S=Surrogate X=Appendix PPRTV E=Based on TEF scheme r=Route Extra Key: C = Cancer endpoint; N = Noncancer endpoint; sat = Saturation Limit; max = Ceiling Limit NDEP Basic Comparison TOXICITY INFORMATION COMPARISON LEVELS LBCLs Indoor Outdoor Levels Skin Industrial/ Industrial/ Commercial Commercial Residential May-17 SFo RfDo IUR RfCi Abs. Residential Worker Worker Ambient Air Water DAF 1 DAF 20 CAS w/o Dermal Chemical Constituents Number 1/(mg/kg-d) (mg/kg-d) (ug/m3)-1 (mg/m3) VOCc Soils Soil (mg/kg) (mg/kg) Soil (mg/kg) (µg/m3) (µg/l) (mg/kg) (mg/kg) Key Key key Key Key Key Key Key Key Acephate 30560-19-1 8.70E-03 I 4.00E-03 I 0.10 5.59E+01 C 7.52E+02 C 2.95E+02 C 7.73E+00 C Acetaldehyde 75-07-0 2.20E-06 I 9.00E-03 I V 1.23E+01 C 5.35E+01 C 1.00E+05 max 1.28E+00 C 2.55E+00 C Acetochlor 34256-82-1 2.00E-02 I 0.10 1.23E+03 N 4.67E+04 N 1.83E+04 N 6.67E+02 N Acetone 67-64-1 9.00E-01 I 3.10E+01 A V 7.04E+04 N 1.00E+05 max 1.00E+05 max 3.23E+04 N 2.05E+04 N 8.00E-01 1.60E+01 Acetone Cyanohydrin 75-86-5 2.00E-03 X 0.10 1.00E+05 max 1.00E+05 max 1.00E+05 max 2.09E+00 N Acetonitrile 75-05-8 6.00E-02 I V 1.00E+05 max 3.75E+03 N 1.00E+05 max 6.26E+01 N 1.25E+02 N Acetophenone 98-86-2 1.00E-01 I V 2.52E+03 sat 2.52E+03 sat 2.52E+03 sat 3.34E+03 N Acetylaminofluorene, 2- 53-96-3 3.80E+00 CA 1.30E-03 CA 0.10 1.28E-01 C 1.72E+00 C 6.75E-01 C 2.16E-03 C 1.77E-02 C Acrolein 107-02-8 5.00E-04 I 2.00E-05 I V -

Toxicity Summary for Antimony December 1992
TOXICITY SUMMARY FOR ANTIMONY DECEMBER 1992 Prepared by Robert A. Young, Ph.D., D.A.B.T. Chemical Hazard Evaluation and Communication Group Biomedical and Environmental Information Analysis Section Health and Safety Research Division Oak Ridge National Laboratory* Oak Ridge, Tennessee Prepared for OAK RIDGE RESERVATION ENVIRONMENTAL RESTORATION PROGRAM *Managed by Martin Marietta Energy Systems, Inc., for the U.S. Department of Energy under Contract No. DE-AC05-84OR21400 This page intentionally left blank. EXECUTIVE SUMMARY Antimony (Sb) is a naturally occurring metal that is used in various manufacturing processes. It exists in valence states of 3 and 5 (Budavari, 1989; ATSDR, 1990). Antimony is a common urban air pollutant (Beliles, 1979). Exposure to antimony may be via inhalation, oral and dermal routes (ATSDR, 1990). Antimony is sparingly absorbed following ingestion or inhalation (Felicetti et al., 1974a; Gerber et al., 1982; ATSDR, 1990). Both gastrointestinal and pulmonary absorption are a function of compound solubility. Antimony is transported in the blood, its distribution varying among species and dependent on its valence state (Felicetti et al., 1974b). Antimony is not metabolized but may bind to macromolecules and react covalently with sulfhydryl and phosphate groups (ATSDR, 1990). Excretion of antimony is primarily via the urine and feces, and is also dependent upon valence state (Cooper et al., 1968; Ludersdorf et al., 1987; ATSDR, 1990). Acute oral exposure of humans and animals to high doses of antimony or antimony-containing compounds (antimonials) may cause gastrointestinal disorders (vomiting, diarrhea), respiratory difficulties, and death at extremely high doses (Bradley and Frederick, 1941; Beliles, 1979; ATSDR, 1990). -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -
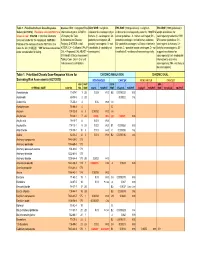
Table 1. Prioritized Chronic Dose-Response Values for Screening Risk Assessments
Table 1. Prioritized Chronic Dose-Response Sources: IRIS = Integrated Risk IARC WOE = weight-of- EPA WOE (1986 guidelines) = weight-of- EPA WOE (1999 guidelines) = Values (4/27/2010). Revisions since 06/12/07 are Information System; ATSDR = evidence for carcinogenicity in evidence for carcinogenicity under the 1986 EPA weight-of-evidence for shown in red. CAS NO. = Chemical Abstracts US Agency for Toxic humans (1 - carcinogenic; 2A - cancer guidelines: A - human carcinogen; B1 - carcinogenicity under the 1999 Services number for the compound. HAP NO. = Substances and Disease probably carcinogenic; 2B - probable carcinogen, limited human evidence; EPA cancer guidelines: CH - Position of the compound on the HAP list in the Registry; D-ATSDR = draft possibly carcinogenic; 3 - not B2 - probable carcinogen, sufficient evidence in carcinogenic to humans; LH - Clean Air Act (112[b][2]). "999" denotes substances ATSDR; CA = California EPA; P- classifiable; 4 - probably not animals; C - possible human carcinogen; D - not likely to be carcinogenic; SE - under consideration for listing. CAL = Proposed CAL; HEAST = carcinogenic). classifiable E - evidence of noncarcinogenicity. suggestive evidence for EPA Health Effects Assessment carcinogenicity; InI - inadequate Tables; Conv. Oral = Oral unit information to determine risk converted to inhalation. carcinogenicity; NH - not likely to be carcinogenic). Table 1. Prioritized Chronic Dose-Response Values for CHRONIC INHALATION CHRONIC ORAL Screening Risk Assessments (4/27/2010) NONCANCER CANCER NONCANCER -

Rpt POL-TOXIC AIR POLLUTANTS 98 BY
SWCAA TOXIC AIR POLLUTANTS '98 by CAS ASIL TAP SQER CAS No HAP POLLUTANT NAME HAP CAT 24hr ug/m3 Ann ug/m3 Class lbs/yr lbs/hr none17 BN 1750 0.20 ALUMINUM compounds none0.00023 AY None None ARSENIC compounds (E649418) ARSENIC COMPOUNDS none0.12 AY 20 None BENZENE, TOLUENE, ETHYLBENZENE, XYLENES BENZENE none0.12 AY 20 None BTEX BENZENE none0.000083 AY None None CHROMIUM (VI) compounds CHROMIUM COMPOUN none0.000083 AY None None CHROMIUM compounds (E649962) CHROMIUM COMPOUN none0.0016 AY 0.5 None COKE OVEN COMPOUNDS (E649830) - CAA 112B COKE OVEN EMISSIONS none3.3 BN 175 0.02 COPPER compounds none0.67 BN 175 0.02 COTTON DUST (raw) none17 BY 1,750 0.20 CYANIDE compounds CYANIDE COMPOUNDS none33 BN 5,250 0.60 FIBROUS GLASS DUST none33 BY 5,250 0.60 FINE MINERAL FIBERS FINE MINERAL FIBERS none8.3 BN 175 0.20 FLUORIDES, as F, containing fluoride, NOS none0.00000003 AY None None FURANS, NITRO- DIOXINS/FURANS none5900 BY 43,748 5.0 HEXANE, other isomers none3.3 BN 175 0.02 IRON SALTS, soluble as Fe none00 AN None None ISOPROPYL OILS none0.5 AY None None LEAD compounds (E650002) LEAD COMPOUNDS none0.4 BY 175 0.02 MANGANESE compounds (E650010) MANGANESE COMPOU none0.33 BY 175 0.02 MERCURY compounds (E650028) MERCURY COMPOUND none33 BY 5,250 0.60 MINERAL FIBERS ((fine), incl glass, glass wool, rock wool, slag w FINE MINERAL FIBERS none0.0021 AY 0.5 None NICKEL 59 (NY059280) NICKEL COMPOUNDS none0.0021 AY 0.5 None NICKEL compounds (E650036) NICKEL COMPOUNDS none0.00000003 AY None None NITROFURANS (nitrofurans furazolidone) DIOXINS/FURANS none0.0013 -

Scientific Basis for Swedish Occupational Standards XXI, Arbete Och Hälsa Vetenskaplig Skriftserie, 2000:22
nr 2000:22 Scientific Basis for Swedish Occupational Standards XXI Criteria Group for Occupational Standards Ed. Johan Montelius National Institute for Working Life S-112 79 Stockholm, Sweden Translation: Frances Van Sant arbete och hälsa | vetenskaplig skriftserie isbn 91-7045-582-1 issn 0346-7821 http://www.niwl.se/ah/ National Institute for Working Life The National Institute for Working Life is Sweden’s national centre for work life research, development and training. The labour market, occupational safety and health, and work organi- sation are our main fields of activity. The creation and use of knowledge through learning, information and documentation are important to the Institute, as is international co-operation. The Institute is collaborating with interested parties in various develop- ment projects. The areas in which the Institute is active include: • labour market and labour law, • work organisation, • musculoskeletal disorders, • chemical substances and allergens, noise and electromagnetic fields, • the psychosocial problems and strain-related disorders in modern working life. ARBETE OCH HÄLSA Editor-in-chief: Staffan Marklund Co-editors: Mikael Bergenheim, Anders Kjellberg, Birgitta Meding, Gunnar Rosén och Ewa Wigaeus Tornqvist © National Institut for Working Life & authors 2000 National Institute for Working Life S-112 79 Stockholm Sweden ISBN 91–7045–582–1 ISSN 0346–7821 http://www.niwl.se/ah/ Printed at CM Gruppen Preface The Criteria Group of the Swedish National Institute for Working Life (NIWL) has the task of gathering and evaluating data which can be used as a scientific basis for the proposal of occupational exposure limits given by the National Board of Occupational Safety and Health (NBOSH). -

Alkali Oxide-Tantalum, Niobium and Antimony Oxide Ionic Conductors, RASA CR-13^869, 76 Pages (National Technical Information Service
- - J HPS' 3/3-6* dfo+t f NBSIR 75-754 NASA CR-1 34869 A Rota, R. 3., Brewer, 3., Parkier, H. 3., Minor, D. B., Waring, J. L., Alkali oxide-tantalum, niobium and antimony oxide ionic conductors, RASA CR-13^869, 76 pages (National Technical Information Service, Springfield, Va . 1975). ALKALI OXIDE-TANTALUM, NIOBIUM AND ANTIMONY OXIDE IONIC CONDUCTORS by R. S. Roth, W. S. Brower. H. S. Parker, D. B. Minor and J. L. Waring prepared for NATIONAL AERONAUTICS AND SPACE ADMINISTRATION CONTRACT C-50821-C U.S. DEPARTMENT OF COMMERCE NATIONAL BUREAU OF STANDARDS WASHINGTON, D. C. 20234 TABLE OF CONTENTS SUMMARY 3 1.0 INTRODUCTION 5 2.0 DISCUSSION OF RESULTS 6 2.1 The System Nb 0 -4Rb 0 : llNb^ 6 2 5 2 2.2 The System Ta 0 -4Rt> 0: llTa^ 6 2 5 2 2.3 The System Sb_0.-NaSbO_ 7 2 4 j 2.3.1 NaSb0 . 8 3 2.3.2 Pyrochlore Solid Solution 8 2.3.3 Polymorphism of Sb^ 10 2.4 The System Sb„0,-KSb0 o 11 2 4 3 2.4.1 Hydroxyl Ion Stabilization of Cubic Potassium Antimonate 11 2.5 The System Sb o0. -NaSbO„-NaF 13 2 4 3 2.5.1 The System NaSbO^NaF 13 2.5.2 The Ternary System 14 2.6 Other Systems 15 2.6.1 Alkali Bismuthates 15 2.6.2 Alkali-Rare Earth Systems 15 2.6.3 Further Studies in the System Nb 0 :KNb0 16 2 5 3 2.7 Pellet Fabrication 16 2.7.1 41K 0:59Nb 0 ("2:3"*, K Nb 0 ) 16 2 2 5 4 6 17 2.7.2 4Rb 0:llNb 0 (11-layer phase) 18 2 2 5 2.7.3 Large Pellets for Ionic Conductivity Measurements ... -

Alchemical Substances
Alchemical substances Cadmia, which was also called Tuttia or Tutty, was probably zinc carbonate. Philosophers' Wool, or nix alba (white snow). Zinc oxide made by burning zinc in air. Called Zinc White and used as a pigment. White vitriol. Zinc Sulphate. Described by Basil Valentine. Made by lixiviating roasted zinc blende (zinc sulphide). Calamine. Zinc carbonate. Corrosive sublimate. Mercuric chloride. first mentioned by Geber, who prepared it by subliming mercury, calcined green vitriol, common salt and nitre. Calomel. Mercurous chloride. Purgative, made by subliming a mixture of mercuric chloride and metallic mercury, triturated in a mortar. This was heated in a iron pot and the crust of calomel formed on the lid was ground to powder and boiled with water to remove the very poisonous mercuric chloride. Cinnabar. Mercuric sulphide. Turpeth mineral. A hydrolysed form of mercuric sulphate. Yellow crystalline powder, described by Basil Valentine. Mercurius praecipitatus. Red mercuric oxide. Described by Geber. Cinnabar or Vermillion. Mercuric sulphide. Mosaic gold. Golden-yellow glistening scales of crystalline stannic sulphide, made by heating a mixture of tin filings, sulphur and salammoniac. Tin salt. Hydrated stannous chloride. Spiritus fumans. Stannic chloride, discovered by Libavius in 1605, through distilling tin with corrosive sublimate. Butter of tin. Hydrated stannic chloride. Galena. Plumbic sulphide. Chief ore of lead. Lead fume. Lead oxide obtained from the flues at lead smelters. Massicot. Yellow powder form of lead monoxide. Litharge. Reddish-yellow crystalline form of lead monoxide, formed by fusing and powdering massicot. Minium or Red Lead. Triplumbic tetroxide. Formed by roasting litharge in air. Scarlet crystalline powder. Naples yellow, or Cassel yellow. -

Common Name: ANTIMONY TRIOXIDE HAZARD SUMMARY IDENTIFICATION REASON for CITATION HOW to DETERMINE IF YOU ARE BEING EXPOSED WORKP
Common Name: ANTIMONY TRIOXIDE CAS Number: 1309-64-4 RTK Substance number: 0149 DOT Number: UN 1549 Date: February 1998 Revision: June 2004 --------------------------------------------------------------------------- --------------------------------------------------------------------------- HAZARD SUMMARY * Antimony Trioxide can affect you when breathed in. * Exposure to hazardous substances should be routinely * Antimony Trioxide should be handled as a evaluated. This may include collecting personal and area CARCINOGEN--WITH EXTREME CAUTION. air samples. You can obtain copies of sampling results * Contact can irritate and may burn the eyes and skin and from your employer. You have a legal right to this cause an itchy skin rash. information under OSHA 1910.1020. * Exposure to Antimony Trioxide can irritate the nose, * If you think you are experiencing any work-related health mouth, throat and lungs causing cough, wheezing and/or problems, see a doctor trained to recognize occupational shortness of breath. diseases. Take this Fact Sheet with you. * Antimony Trioxide can cause headaches, poor appetite, nausea, vomiting, dry throat, and loss of sleep. WORKPLACE EXPOSURE LIMITS * Prolonged or repeated contact can cause ulcers or sores in The following exposure limits are for Antimony and the nose. compounds (measured as Antimony): * Repeated exposure can affect the lungs and cause an abnormal chest x-ray to develop. OSHA: The legal airborne permissible exposure limit 3 * Antimony Trioxide may damage the kidneys, liver and (PEL) is 0.5 mg/m averaged over an 8-hour heart. workshift. IDENTIFICATION NIOSH: The recommended airborne exposure limit is 0.5 mg/m3 averaged over a 10-hour workshift. Antimony Trioxide is a white, odorless crystalline (sand-like) powder. It is used as a flame-proofing agent, in pigments and ACGIH: The recommended airborne exposure limit is ceramics, to stain iron and copper, and to decolorize glass.