Dependent Modeling Approach Derived from Semi-Empirical Quantum Mechanical Calculations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Retention Indices for Frequently Reported Compounds of Plant Essential Oils
Retention Indices for Frequently Reported Compounds of Plant Essential Oils V. I. Babushok,a) P. J. Linstrom, and I. G. Zenkevichb) National Institute of Standards and Technology, Gaithersburg, Maryland 20899, USA (Received 1 August 2011; accepted 27 September 2011; published online 29 November 2011) Gas chromatographic retention indices were evaluated for 505 frequently reported plant essential oil components using a large retention index database. Retention data are presented for three types of commonly used stationary phases: dimethyl silicone (nonpolar), dimethyl sili- cone with 5% phenyl groups (slightly polar), and polyethylene glycol (polar) stationary phases. The evaluations are based on the treatment of multiple measurements with the number of data records ranging from about 5 to 800 per compound. Data analysis was limited to temperature programmed conditions. The data reported include the average and median values of retention index with standard deviations and confidence intervals. VC 2011 by the U.S. Secretary of Commerce on behalf of the United States. All rights reserved. [doi:10.1063/1.3653552] Key words: essential oils; gas chromatography; Kova´ts indices; linear indices; retention indices; identification; flavor; olfaction. CONTENTS 1. Introduction The practical applications of plant essential oils are very 1. Introduction................................ 1 diverse. They are used for the production of food, drugs, per- fumes, aromatherapy, and many other applications.1–4 The 2. Retention Indices ........................... 2 need for identification of essential oil components ranges 3. Retention Data Presentation and Discussion . 2 from product quality control to basic research. The identifi- 4. Summary.................................. 45 cation of unknown compounds remains a complex problem, in spite of great progress made in analytical techniques over 5. -

Effect of Enzymes on Strawberry Volatiles During Storage, at Different Ripeness
Effect of Enzymes on Strawberry Volatiles During Storage, at Different Ripeness Level, in Different Cultivars and During Eating Thesis Presented in Partial Fulfillment of the Requirements for the Degree Master of Science in the Graduate School of The Ohio State University By Gulsah Ozcan Graduate Program in Food Science and Technology The Ohio State University 2010 Thesis Committee: Sheryl Ann Barringer, Adviser W. James Harper John Litchfield 1 Copyright by Gülşah Özcan 2010 ii ABSTRACT Strawberry samples with enzyme activity and without enzyme activity (stannous chloride added) were measured for real time formation of lipoxygenase (LOX) derived aroma compounds after 5 min pureeing using selected ion flow tube mass spectrometry (SIFT-MS). The concentration of (Z)-3-hexenal and (E)-2-hexenal increased immediately after blending and gradually decreased over time while hexanal concentration increased for at least 5 min in ground strawberries. The formation of hexanal was slower than the formation of (Z)-3-hexenal and (E)-2-hexenal in the headspace of pureed strawberries. The concentration of LOX aldehydes and esters significantly increased during refrigerated storage. Damaging strawberries increased the concentration of LOX aldehydes but did not significantly affect the concentration of esters. The concentrations of many of the esters were strongly correlated to their corresponded acids and/or aldehydes. The concentration of LOX generated aldehydes decreased during ripening, while fruity esters increased. Different varieties had different aroma profiles and esters were the greatest percentage of the volatiles. The aroma release of some of the LOX derived aldehydes in the mouthspace in whole strawberries compared to chopped strawberries showed that these volatiles are formed in the mouth during chewing. -

Ethers, Ether-Alcohols, Ether-Phenols, Ether
29-IV Sub-Chapter IV ETHERS, ALCOHOL PEROXIDES, ETHER PEROXIDES, KETONE PEROXIDES, EPOXIDES WITH A THREE-MEMBERED RING, ACETALS AND HEMIACETALS, AND THEIR HALOGENATED, SULPHONATED, NITRATED OR NITROSATED DERIVATIVES 29.09 - Ethers, ether-alcohols, ether-phenols, ether-alcohol-phenols, alcohol peroxides, ether peroxides, ketone peroxides (whether or not chemically defined), and their halogenated, sulphonated, nitrated or nitrosated derivatives. - Acyclic ethers and their halogenated, sulphonated, nitrated or nitrosated derivatives : 2909.11 - - Diethyl ether 2909.19 - - Other 2909.20 - Cyclanic, cyclenic or cycloterpenic ethers and their halogenated, sulphonated, nitrated or nitrosated derivatives 2909.30 - Aromatic ethers and their halogenated, sulphonated, nitrated or nitrosated derivatives - Ether-alcohols and their halogenated, sulphonated, nitrated or nitrosated derivatives : 2909.41 - - 2,2’-Oxydiethanol (diethylene glycol, digol) 2909.43 - - Monobutyl ethers of ethylene glycol or of diethylene glycol 2909.44 - - Other monoalkylethers of ethylene glycol or of diethylene glycol 2909.49 - - Other 2909.50 - Ether-phenols, ether-alcohol-phenols and their halogenated, sulphonated, nitrated or nitrosated derivatives 2909.60 - Alcohol peroxides, ether peroxides, ketone peroxides and their halogenated, sulphonated, nitrated or nitrosated derivatives (A) ETHERS Ethers may be considered as alcohols or phenols in which the hydrogen atom of the hydroxyl group is replaced by a hydrocarbon radical (alkyl or aryl). They have the general formula : (R-O-R1), where R and R1 may be the same or different. These ethers are very stable, neutral substances. If the radicals belong to the acyclic series, the ether is also acyclic; cyclic radicals give cyclic ethers. The first ether in the acyclic series is gaseous, but others are volatile liquids with a characteristic odour of ether; the higher members are liquids or sometimes solids. -

Bioelectronic Nose Based on Single-Stranded DNA And
nanomaterials Article Bioelectronic Nose Based on Single-Stranded DNA and Single-Walled Carbon Nanotube to Identify a Major Plant Volatile Organic Compound (p-Ethylphenol) Released by Phytophthora Cactorum Infected Strawberries Hui Wang 1,2,* , Yue Wang 1, Xiaopeng Hou 2 and Benhai Xiong 1,* 1 State Key Laboratory of Animal Nutrition, Institute of Animal Sciences, Chinese Academy of Agricultural Sciences, Beijing 100193, China; [email protected] 2 Research Institute of Wood Industry, Chinese Academy of Forestry, Beijing 100091, China; [email protected] * Correspondence: [email protected] or [email protected] (H.W.); [email protected] (B.X.); Tel.: +86-010-62811680 (B.X.) Received: 5 February 2020; Accepted: 3 March 2020; Published: 7 March 2020 Abstract: The metabolic activity in plants or fruits is associated with volatile organic compounds (VOCs), which can help identify the different diseases. P-ethylphenol has been demonstrated as one of the most important VOCs released by the Phytophthora cactorum (P. cactorum) infected strawberries. In this study, a bioelectronic nose based on a gas biosensor array and signal processing model was developed for the noninvasive diagnostics of the P.cactorum infected strawberries, which could overcome the limitations of the traditional spectral analysis methods. The gas biosensor array was fabricated using the single-wall carbon nanotubes (SWNTs) immobilized on the surface of field-effect transistor, and then non-covalently functionalized with different single-strand DNAs (ssDNA) through π–π interaction. The characteristics of ssDNA-SWNTs were investigated using scanning electron microscope, atomic force microscopy, Raman, UV spectroscopy, and electrical measurements, indicating that ssDNA-SWNTs revealed excellent stability and repeatability. -

ECO-Ssls for Pahs
Ecological Soil Screening Levels for Polycyclic Aromatic Hydrocarbons (PAHs) Interim Final OSWER Directive 9285.7-78 U.S. Environmental Protection Agency Office of Solid Waste and Emergency Response 1200 Pennsylvania Avenue, N.W. Washington, DC 20460 June 2007 This page intentionally left blank TABLE OF CONTENTS 1.0 INTRODUCTION .......................................................1 2.0 SUMMARY OF ECO-SSLs FOR PAHs......................................1 3.0 ECO-SSL FOR TERRESTRIAL PLANTS....................................4 5.0 ECO-SSL FOR AVIAN WILDLIFE.........................................8 6.0 ECO-SSL FOR MAMMALIAN WILDLIFE..................................8 6.1 Mammalian TRV ...................................................8 6.2 Estimation of Dose and Calculation of the Eco-SSL ........................9 7.0 REFERENCES .........................................................16 7.1 General PAH References ............................................16 7.2 References Used for Derivation of Plant and Soil Invertebrate Eco-SSLs ......17 7.3 References Rejected for Use in Derivation of Plant and Soil Invertebrate Eco-SSLs ...............................................................18 7.4 References Used in Derivation of Wildlife TRVs .........................25 7.5 References Rejected for Use in Derivation of Wildlife TRV ................28 i LIST OF TABLES Table 2.1 PAH Eco-SSLs (mg/kg dry weight in soil) ..............................4 Table 3.1 Plant Toxicity Data - PAHs ..........................................5 Table 4.1 -

Original Paper Comparison of Volatile Compounds in 'Fuji' Apples in The
_ Food Science and Technology Research, 23 (1), 79 89, 2017 Copyright © 2017, Japanese Society for Food Science and Technology doi: 10.3136/fstr.23.79 http://www.jsfst.or.jp Original paper Comparison of Volatile Compounds in ‘Fuji’ Apples in the Different Regions in China 1 2* 3 2 2 2 Ling QIN , Qin-Ping WEI , Wen-Huai KANG , Qiang ZHANG , Jian SUN and Song-Zhong LIU 1Department of Life Science, Hebei Normal University of Science and Technology, Qinhuangdao 066004 P. R. China 2Institute of Forestry & Pomology, Beijing Academy of Agriculture & Forestry Sciences,Beijing 100093 P.R. China 3College of Bioscience and Bioengineering, Hebei University of Science and Technology, Shijiazhuang 050018 P.R. China Received May 25, 2015 ; Accepted September 28, 2016 The characteristics of the volatiles from 43 ‘Fuji’ apples representing 14 different apple production regions in China were investigated using headspace-solid phase micro-extraction (HS-SPME) combined with gas chromatography–mass spectrometry (GC-MS). The results obtained from this experiment showed that sixty- four volatile compounds were identified in ‘Fuji’ apples collected from 43 counties in China. The major volatile compounds were identified as 2-methyl butyl acetate and hexyl acetate. The composition of volatiles and their contents in ‘Fuji’ apples varied in different regions. All of the ‘Fuji’ apple samples could be classified into the following groups using a principal component analysis of the volatiles: (1) apples with high concentrations of hexyl acetate and (Z)-3-hexenyl acetate, -

Handbook of Physical-Chemical Properties and Environmental Fate for Organic Chemicals.--2Nd Ed
Second Edition Physical-Chemical Properties and Environmental Fate for Organic Chemicals Volume I Introduction and Hydrocarbons Volume II Halogenated Hydrocarbons Volume III HANDBOOK OF HANDBOOK Oxygen Containing Compounds Volume IV Nitrogen and Sulfur Containing Compounds and Pesticides © 2006 by Taylor & Francis Group, LLC Second Edition Physical-Chemical Properties and Environmental Fate for Organic Chemicals Volume I Introduction and Hydrocarbons Volume II Halogenated Hydrocarbons Volume III Oxygen Containing Compounds HANDBOOK OF HANDBOOK Volume IV Nitrogen and Sulfur Containing Compounds and Pesticides Donald Mackay Wan Ying Shiu Kuo-Ching Ma Sum Chi Lee Boca Raton London New York A CRC title, part of the Taylor & Francis imprint, a member of the Taylor & Francis Group, the academic division of T&F Informa plc. © 2006 by Taylor & Francis Group, LLC Published in 2006 by CRC Press Taylor & Francis Group 6000 Broken Sound Parkway NW, Suite 300 Boca Raton, FL 33487-2742 © 2006 by Taylor & Francis Group, LLC CRC Press is an imprint of Taylor & Francis Group No claim to original U.S. Government works Printed in the United States of America on acid-free paper 10987654321 International Standard Book Number-10: 1-56670-687-4 (Hardcover) International Standard Book Number-13: 978-1-56670-687-2 (Hardcover) Library of Congress Card Number 2005051402 This book contains information obtained from authentic and highly regarded sources. Reprinted material is quoted with permission, and sources are indicated. A wide variety of references are listed. Reasonable efforts have been made to publish reliable data and information, but the author and the publisher cannot assume responsibility for the validity of all materials or for the consequences of their use. -

Expanding the Modular Ester Fermentative Pathways for Combinatorial Biosynthesis of Esters from Volatile Organic Acids
ARTICLE Expanding the Modular Ester Fermentative Pathways for Combinatorial Biosynthesis of Esters From Volatile Organic Acids Donovan S. Layton,1,2 Cong T. Trinh1,2,3 1 Department of Chemical and Biomolecular Engineering, University of Tennessee, Knoxville, Tennessee 2 BioEnergy Science Center (BESC), Oak Ridge National Laboratory, Oak Ridge, Tennessee 3 Bredesen Center for Interdisciplinary Research and Graduate Education, University of Tennessee, Knoxville, Tennessee; telephone: þ865-974-8121; fax: 865-974-7076; e-mail: [email protected] Biotechnol. Bioeng. 2016;113: 1764–1776. ABSTRACT: Volatile organic acids are byproducts of fermentative ß 2016 Wiley Periodicals, Inc. metabolism, for example, anaerobic digestion of lignocellulosic KEYWORDS: modular chassis cell; carboxylate; ester; acyl acetate; biomass or organic wastes, and are often times undesired inhibiting acyl acylate; ester fermentative pathway cell growth and reducing directed formation of the desired products. Here, we devised a general framework for upgrading these volatile organic acids to high-value esters that can be used as flavors, fragrances, solvents, and biofuels. This framework employs the acid-to-ester modules, consisting of an AAT (alcohol Introduction acyltransferase) plus ACT (acyl CoA transferase) submodule and an alcohol submodule, for co-fermentation of sugars and organic Harnessing renewable or waste feedstocks (e.g., switchgrass, corn acids to acyl CoAs and alcohols to form a combinatorial library of stover, agricultural residue, or municipal solid waste) -
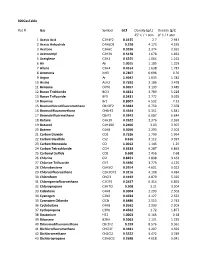
Gas Conversion Factor for 300 Series
300GasTable Rec # Gas Symbol GCF Density (g/L) Density (g/L) 25° C / 1 atm 0° C / 1 atm 1 Acetic Acid C2H4F2 0.4155 2.7 2.947 2 Acetic Anhydride C4H6O3 0.258 4.173 4.555 3 Acetone C3H6O 0.3556 2.374 2.591 4 Acetonitryl C2H3N 0.5178 1.678 1.832 5 Acetylene C2H2 0.6255 1.064 1.162 6 Air Air 1.0015 1.185 1.293 7 Allene C3H4 0.4514 1.638 1.787 8 Ammonia NH3 0.7807 0.696 0.76 9 Argon Ar 1.4047 1.633 1.782 10 Arsine AsH3 0.7592 3.186 3.478 11 Benzene C6H6 0.3057 3.193 3.485 12 Boron Trichloride BCl3 0.4421 4.789 5.228 13 Boron Triflouride BF3 0.5431 2.772 3.025 14 Bromine Br2 0.8007 6.532 7.13 15 Bromochlorodifluoromethane CBrClF2 0.3684 6.759 7.378 16 Bromodifluoromethane CHBrF2 0.4644 5.351 5.841 17 Bromotrifluormethane CBrF3 0.3943 6.087 6.644 18 Butane C4H10 0.2622 2.376 2.593 19 Butanol C4H10O 0.2406 3.03 3.307 20 Butene C4H8 0.3056 2.293 2.503 21 Carbon Dioxide CO2 0.7526 1.799 1.964 22 Carbon Disulfide CS2 0.616 3.112 3.397 23 Carbon Monoxide CO 1.0012 1.145 1.25 24 Carbon Tetrachloride CCl4 0.3333 6.287 6.863 25 Carbonyl Sulfide COS 0.668 2.456 2.68 26 Chlorine Cl2 0.8451 2.898 3.163 27 Chlorine Trifluoride ClF3 0.4496 3.779 4.125 28 Chlorobenzene C6H5Cl 0.2614 4.601 5.022 29 Chlorodifluoroethane C2H3ClF2 0.3216 4.108 4.484 30 Chloroform CHCl3 0.4192 4.879 5.326 31 Chloropentafluoroethane C2ClF5 0.2437 6.314 6.892 32 Chloropropane C3H7Cl 0.308 3.21 3.504 33 Cisbutene C4H8 0.3004 2.293 2.503 34 Cyanogen C2N2 0.4924 2.127 2.322 35 Cyanogen Chloride ClCN 0.6486 2.513 2.743 36 Cyclobutane C4H8 0.3562 2.293 2.503 37 Cyclopropane C3H6 0.4562 -

Japanese Flavoring Agents As Food Additives
Japanese Flavoring Agents as Food Additives In Japan, synthetic flavoring agents are allowed to be used only when they are designated by the Minister of Health, Labour and Welfare as food additives under the Japanese Food Sanitation Act. Currently, we have identified 170 chemical substances which are commonly used as flavorings as shown in Table 1. Table 2 lists 18 groups which are also from the official list of “designated additives” and contain 3004 additional flavor materials. Each of the 18 groups in Table 2 contains substances that are similar in chemical structure. For links to a complete listing of the Japanese additives used in food: (a) Designated additives, (b) Existing food additives, (c) Natural flavoring agents and (d) Ordinary foods used as food additives - go to http://www.mhlw.go.jp/english/topics/foodsafety/foodadditives/index.html Check for Flavoring updates at http://www.jffma-jp.org/english/information.html Provided with Updated Revisions (as of April 2015) By Leffingwell & Associates Table 1. Designated additives used as flavoring substances Compound Synonym or Old name CAS Acesulfame Potassium 55589-62-3 Acetaldehyde (New as of 2006.05.16) ethanal 75-07-0 Acetophenone acetophenone 98-86-2 Acetic acid, Glacial 64-19-7 Adipic Acid 124-04-9 714229-20-6 Advantame (New as of 2015.02.20) 245650-17-3 DL-Alanine 302-72-7 Allyl cyclohexylpropionate allyl cyclohexanepropionate 2705-87-5 Allyl hexanoate allyl hexanoate 123-68-2 Allyl isothiocyanate allyl isothiocyanate 57503 (3-Amino-3-carboxypropyl)dimethylsulfonium chloride -

And Perfluoroalkyl Substances (PFAS): Sources, Pathways and Environmental Data
Poly- and perfluoroalkyl substances (PFAS): sources, pathways and environmental data Chief Scientist’s Group report August 2021 We are the Environment Agency. We protect and improve the environment. We help people and wildlife adapt to climate change and reduce its impacts, including flooding, drought, sea level rise and coastal erosion. We improve the quality of our water, land and air by tackling pollution. We work with businesses to help them comply with environmental regulations. A healthy and diverse environment enhances people's lives and contributes to economic growth. We can’t do this alone. We work as part of the Defra group (Department for Environment, Food & Rural Affairs), with the rest of government, local councils, businesses, civil society groups and local communities to create a better place for people and wildlife. Published by: Author: Emma Pemberton Environment Agency Horizon House, Deanery Road, Environment Agency’s Project Manager: Bristol BS1 5AH Mark Sinton www.gov.uk/environment-agency Citation: Environment Agency (2021) Poly- and © Environment Agency 2021 perfluoroalkyl substances (PFAS): sources, pathways and environmental All rights reserved. This document may data. Environment Agency, Bri be reproduced with prior permission of the Environment Agency. Further copies of this report are available from our publications catalogue: www.gov.uk/government/publications or our National Customer Contact Centre: 03708 506 506 Email: research@environment- agency.gov.uk 2 of 110 Research at the Environment Agency Scientific research and analysis underpins everything the Environment Agency does. It helps us to understand and manage the environment effectively. Our own experts work with leading scientific organisations, universities and other parts of the Defra group to bring the best knowledge to bear on the environmental problems that we face now and in the future. -

Reuse of Oak Chips for Modification of the Volatile Fraction of Alcoholic Beverages
LWT - Food Science and Technology 135 (2021) 110046 Contents lists available at ScienceDirect LWT journal homepage: www.elsevier.com/locate/lwt Reuse of oak chips for modification of the volatile fraction of alcoholic beverages Eduardo Coelho *, Jos´e A. Teixeira , Teresa Tavares , Lucília Domingues , Jos´e M. Oliveira CEB – Centre of Biological Engineering, University of Minho, 4710-057, Braga, Portugal ARTICLE INFO ABSTRACT Keywords: New or used barrels can be applied in ageing of alcoholic beverages. Compounds adsorbed in wood migrate Wood ageing between beverages along with wood extractives. As barrel ageing is costly and time-consuming, processes using Barrel alternatives wood fragments have been gaining interest. These generate wood residues for which the reuse is still not well Volatile compounds established. This work aims at the reuse of oak fragments for the additive ageing of alcoholic beverages. Oak Sensory properties chips, previously immersed in fortifiedwine, were applied to beer, wine and grape marc spirit. Wood compounds and adsorbed wine volatiles were extracted, with more impact and satisfactory yields on beer composition. Also, wood adsorbed beverages compounds in a subtractive ageing phenomena. Beer formulations using different binomial wood concentration/temperature combinations were generated and presented to trained tasters. Higher temperatures and wood concentrations led to prominence of wood descriptors and lower perception of fruity and floral aromas, reflecting the changes in chemical composition. 1. Introduction several other (Mosedale & Puech, 2003). During its lifecycle, cooperage wood can be used, reused, regenerated and eventually discarded. For Wood ageing is commonly used as a strategy to modify and enhance barrels, there is a well-established reuse flow depending on the aged the composition of alcoholic beverages.