List Item Synagis-H-C-257-P46-0036 : EPAR
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Phage Display-Based Strategies for Cloning and Optimization of Monoclonal Antibodies Directed Against Human Pathogens
Int. J. Mol. Sci. 2012, 13, 8273-8292; doi:10.3390/ijms13078273 OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms Review Phage Display-based Strategies for Cloning and Optimization of Monoclonal Antibodies Directed against Human Pathogens Nicola Clementi *,†, Nicasio Mancini †, Laura Solforosi, Matteo Castelli, Massimo Clementi and Roberto Burioni Microbiology and Virology Unit, “Vita-Salute” San Raffaele University, Milan 20132, Italy; E-Mails: [email protected] (N.M.); [email protected] (L.S.); [email protected] (M.C.); [email protected] (M.C.); [email protected] (R.B.) † These authors contributed equally to this work. * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +39-2-2643-5082; Fax: +39-2-2643-4288. Received: 16 March 2012; in revised form: 25 June 2012 / Accepted: 27 June 2012 / Published: 4 July 2012 Abstract: In the last two decades, several phage display-selected monoclonal antibodies (mAbs) have been described in the literature and a few of them have managed to reach the clinics. Among these, the anti-respiratory syncytial virus (RSV) Palivizumab, a phage-display optimized mAb, is the only marketed mAb directed against microbial pathogens. Palivizumab is a clear example of the importance of choosing the most appropriate strategy when selecting or optimizing an anti-infectious mAb. From this perspective, the extreme versatility of phage-display technology makes it a useful tool when setting up different strategies for the selection of mAbs directed against human pathogens, especially when their possible clinical use is considered. -

Pharmacokinetic-Pharmacodynamic Modelling of Systemic IL13 Blockade by Monoclonal Antibody Therapy: a Free Assay Disguised As Total
pharmaceutics Article Pharmacokinetic-Pharmacodynamic Modelling of Systemic IL13 Blockade by Monoclonal Antibody Therapy: A Free Assay Disguised as Total John Hood 1,*, Ignacio González-García 1 , Nicholas White 1, Leeron Marshall 1,2, Vincent F. S. Dubois 1 , Paolo Vicini 1,3 and Paul G. Baverel 1,4 1 Clinical Pharmacology and Quantitative Pharmacology, AstraZeneca, Cambridge CB21 6GH, UK; [email protected] (I.G.-G.); [email protected] (N.W.); [email protected] (L.M.); [email protected] (V.F.S.D.); [email protected] (P.V.); [email protected] (P.G.B.) 2 Salford Royal Foundation Trust, Salford M6 8HD, UK 3 Confo Therapeutics, 9052 Ghent, Zwijnaarde, Belgium 4 Roche Pharma Research and Early Development, Clinical Pharmacology, Pharmaceutical Sciences, Roche Innovation Center Basel F. Hoffmann-La Roche Ltd., CH-4070 Basel, Switzerland * Correspondence: [email protected]; Tel.: +44-1223-749-6288 Abstract: A sequential pharmacokinetic (PK) and pharmacodynamic (PD) model was built with Nonlinear Mixed Effects Modelling based on data from a first-in-human trial of a novel biologic, MEDI7836. MEDI7836 is a human immunoglobulin G1 lambda (IgG1λ-YTE) monoclonal antibody, Citation: Hood, J.; González-García, with an Fc modification to reduce metabolic clearance. MEDI7836 specifically binds to, and function- I.; White, N.; Marshall, L.; Dubois, ally neutralizes interleukin-13. Thirty-two healthy male adults were enrolled into a dose-escalation V.F.S.; Vicini, P.; Baverel, P.G. clinical trial. Four active doses were tested (30, 105, 300, and 600 mg) with 6 volunteers enrolled Pharmacokinetic-Pharmacodynamic per cohort. Eight volunteers received placebo as control. -

Structural Basis for Nonneutralizing Antibody Competition at Antigenic
Structural basis for nonneutralizing antibody PNAS PLUS competition at antigenic site II of the respiratory syncytial virus fusion protein Jarrod J. Mousaa, Marion F. Sauerb, Alexander M. Sevyb, Jessica A. Finnc, John T. Batesa, Gabriela Alvaradoc, Hannah G. Kinga, Leah B. Loerinca, Rachel H. Fongd, Benjamin J. Doranzd, Bruno E. Correiae, Oleksandr Kalyuzhniye, Xiaolin Wenf, Theodore S. Jardetzkyf, William R. Schiefe, Melanie D. Ohig, Jens Meilerh, and James E. Crowe Jr.a,c,i,1 aVanderbilt Vaccine Center, Vanderbilt University Medical Center, Nashville, TN 37232; bChemical and Physical Biology Program, Vanderbilt University Medical Center, Nashville, TN 37232; cDepartment of Pathology, Microbiology, and Immunology, Vanderbilt University, Nashville, TN 37232; dIntegral Molecular, Inc., Philadelphia, PA 19104; eDepartment of Immunology and Microbial Science, The Scripps Research Institute, La Jolla, CA 92037; fDepartment of Structural Biology, Stanford University School of Medicine, Stanford, CA 94305; gDepartment of Cell and Developmental Biology, Vanderbilt University Medical Center, Nashville, TN 37232; hDepartment of Chemistry, Vanderbilt University, Nashville, TN 37232; and iDepartment of Pediatrics, Vanderbilt University School of Medicine, Nashville, TN 37232 Edited by Rino Rappuoli, GSK Vaccines, Siena, Italy, and approved September 20, 2016 (received for review June 10, 2016) Palivizumab was the first antiviral monoclonal antibody (mAb) rearrangement, resulting in fusion of the viral and cell mem- approved for therapeutic use in -

Motavizumab for Prophylaxis of Respiratory Syncytial Virus in High-Risk Children: a Noninferiority Trial
ARTICLES Motavizumab for Prophylaxis of Respiratory Syncytial Virus in High-Risk Children: A Noninferiority Trial AUTHORS: Xavier Carbonell-Estrany, MD, PhD,a Eric A. F. WHAT’S KNOWN ON THIS SUBJECT: Monthly prophylaxis with Simo˜es, MD, MB, BS, DCH,b,c Ron Dagan, MD,d Caroline B. palivizumab has been shown to reduce RSV hospitalizations by Hall, MD,e Brian Harris, MS,f Micki Hultquist, MS,f Edward ϳ50% overall compared with placebo in children at high risk for f f M. Connor, MD, and Genevieve A. Losonsky, MD, for the severe RSV disease. Motavizumab, a monoclonal antibody Motavizumab Study Group developed from palivizumab, has enhanced preclinical activity aNeonatology Service, Hospital Clínic, Agrupacio´Sanite`ria Clínic, against RSV. Hospital de Sant Joan de De´u, Barcelona, Spain; bDepartment of Pediatrics, University of Colorado School of Medicine, Denver, Colorado; cDivision of Infectious Diseases, Children’s Hospital, WHAT THIS STUDY ADDS: Motavizumab may offer an improved Denver, Colorado; dPediatric Infectious Disease Unit, Soroka alternative in prophylaxis for serious RSV disease in children at University Medical Center and Faculty of Health Sciences, Ben- high risk. Motavizumab was noninferior to palivizumab for Gurion University, Beer-Sheva, Israel; eDepartments of Pediatrics prevention of RSV hospitalization (primary end point) and and Medicine, University of Rochester Medical Center, superior to palivizumab for reduction of RSV-specific outpatient Rochester, New York; and fMedImmune, Gaithersburg, Maryland MALRI (a secondary end point). KEY WORDS clinical trial, motavizumab, palivizumab, pediatric, respiratory infection, respiratory syncytial virus ABBREVIATIONS RSV—respiratory syncytial virus CLD—chronic lung disease of prematurity abstract MALRI—medically attended lower respiratory tract infection OBJECTIVE: Palivizumab reduces respiratory syncytial virus (RSV) hospi- OM—otitis media ϳ AE—adverse event talization in children at high risk by 50% compared with placebo. -

(INN) for Biological and Biotechnological Substances
INN Working Document 05.179 Update 2013 International Nonproprietary Names (INN) for biological and biotechnological substances (a review) INN Working Document 05.179 Distr.: GENERAL ENGLISH ONLY 2013 International Nonproprietary Names (INN) for biological and biotechnological substances (a review) International Nonproprietary Names (INN) Programme Technologies Standards and Norms (TSN) Regulation of Medicines and other Health Technologies (RHT) Essential Medicines and Health Products (EMP) International Nonproprietary Names (INN) for biological and biotechnological substances (a review) © World Health Organization 2013 All rights reserved. Publications of the World Health Organization are available on the WHO web site (www.who.int ) or can be purchased from WHO Press, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel.: +41 22 791 3264; fax: +41 22 791 4857; e-mail: [email protected] ). Requests for permission to reproduce or translate WHO publications – whether for sale or for non-commercial distribution – should be addressed to WHO Press through the WHO web site (http://www.who.int/about/licensing/copyright_form/en/index.html ). The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate border lines for which there may not yet be full agreement. The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. -

Michigan Care Improvement Registry HL7 2.5.1 Specification Guide for Query by Parameter (QBP)
Michigan Care Improvement Registry HL7 2.5.1 Specification Guide for Query by Parameter (QBP) Version 1.5 JUNE 7, 2016 Document Description This guide is intended for immunization providers and their vendors to assist in connecting to the Michigan Care Improvement Registry (MCIR). MCIR is an immunization registry that compiles complete immunization histories for children and adults in Michigan. This document explains the technical details of how to Query MCIR for a patient records and receive the immunization history and forecast for next dose due for that patient. Message types supported: QBP Z44^CDC RSP Z42^CDC ACK Message formats supported: HL7 2.5.1 version 1.5 Revision history Revision Date Author Release 1.0 August 1, 2013 Therese Hoyle Release 1.1 January 7,2014 Therese Hoyle Release 1.5 June 7, 2016 Sallie Sims A list of changes may be found at the end of Implementation Guide 1 Michigan Care Improvement Registry Introduction and History MCIR was created in 1998 to collect reliable immunization information and make it accessible to authorized users online. In 2006, MCIR was expanded to include adults. By state law, providers are required to submit childhood immunizations within 72 hours of administration. In addition, providers are allowed and highly encouraged to report adult vaccinations. MCIR benefits health care organizations, schools, licensed childcare programs, and Michigan’s citizens by consolidating immunization information from multiple providers. This reduces vaccine-preventable diseases, over-vaccination, and allows providers to see up-to-date patient immunization history. MCIR also has the ability to assist with pandemic flu preparedness and can track vaccines and medications during a public health emergency. -

Pediatric Pharmacy Practice Making a Difference When and Where It Counts!
A Peer Review Publication of the California Pharmacists Association Vol. LV, No.1 Winter 2008 Pediatric Pharmacy Practice Making a Difference When and Where it Counts! In This Issue: MTM for Pediatric Asthma RSV Season is Here! Esprit Trial 50 Years of Pharmacy Education Special Inserts: 2007 Annual Report Policy Reports FROM THE EDITOR EXECUTIVE EDITOR Lynn W. Rolston MANAGING EDITOR Cathi Lord California PharmacistPharmacy TodayCalifornia Pharmacist EDITORS/PROOFREADERS W Vic Boisseree, Leslie Bailey EDITORIAL REVIEW COMMITTEE Craig Stern, Chair Marshal Abdullah, Student Representative Colleen Barnes, Student Representative Sarica Cohen, Los Angeles Karl Hess, Los Angeles Hilda Ilanjian, Los Angeles Lisa Johnson, Manhattan Beach Lee Klevens, Los Angeles Polina Litmanovich, Los Angeles Quynhlam Tran, Orange County Debbie Veale, Fullerton Graphic Design Creative Directions CALIFORNIA PHARMACISTS ASSOCIATION BOARD OF TRUSTEES Paul W. Lofholm, President Jeffery Goad, President-Elect Gerald Mazzucca, Past President Lynn Rolston, Secretary Kenneth Scott, Treasurer Eric Gupta, Speaker Mark Gilbert, Trustee-At-Large Merilyn Ross, Region 1 Trustee Ed Sherman, Region 2 Trustee Dana Nelson, Region 3 Trustee William Young, Region 4 Trustee Mark Gilbert, Region 5 Trustee Rebecca Cupp, Region 6 Trustee Kathleen Besinque, Region 7 Trustee Steve Gray, Region 8 Trustee Nancy DeGuire, APS Trustee Shirley Fender, ALTC Trustee Candace Fong, AHP Trustee Mark Raus, AEP Trustee Patrick Robinson, AMC Trustee Daniel Wills, APT Trustee Dennis Witherwax, APO Trustee -

(INN) for Biological and Biotechnological Substances
WHO/EMP/RHT/TSN/2019.1 International Nonproprietary Names (INN) for biological and biotechnological substances (a review) 2019 WHO/EMP/RHT/TSN/2019.1 International Nonproprietary Names (INN) for biological and biotechnological substances (a review) 2019 International Nonproprietary Names (INN) Programme Technologies Standards and Norms (TSN) Regulation of Medicines and other Health Technologies (RHT) Essential Medicines and Health Products (EMP) International Nonproprietary Names (INN) for biological and biotechnological substances (a review) FORMER DOCUMENT NUMBER: INN Working Document 05.179 © World Health Organization 2019 All rights reserved. Publications of the World Health Organization are available on the WHO website (www.who.int) or can be purchased from WHO Press, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel.: +41 22 791 3264; fax: +41 22 791 4857; e-mail: [email protected]). Requests for permission to reproduce or translate WHO publications –whether for sale or for non-commercial distribution– should be addressed to WHO Press through the WHO website (www.who.int/about/licensing/copyright_form/en/index.html). The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted and dashed lines on maps represent approximate border lines for which there may not yet be full agreement. The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. -

(RSV F) to Elicit High Neutralizing Antibody Titers
Structural basis for immunization with postfusion respiratory syncytial virus fusion F glycoprotein (RSV F) to elicit high neutralizing antibody titers Kurt A. Swanson1, Ethan C. Settembre1, Christine A. Shaw, Antu K. Dey, Rino Rappuoli2, Christian W. Mandl, Philip R. Dormitzer, and Andrea Carfi2 Novartis Vaccines and Diagnostics, Cambridge, MA 02139 Contributed by Rino Rappuoli, April 26, 2011 (sent for review April 11, 2011) Respiratory syncytial virus (RSV), the main cause of infant bronchio- The prefusion and postfusion forms of RSV F each have potential litis, remains a major unmet vaccine need despite more than 40 shortcomings as vaccine antigens. Large structural differences be- years of vaccine research. Vaccine candidates based on a chief RSV tween the lollipop-shaped prefusion F trimer and the crutch-shaped neutralization antigen, the fusion (F) glycoprotein, have foundered postfusion F trimer are apparent even at the resolution of electron due to problems with stability, purity, reproducibility, and potency. microscopy of negatively stained specimens, suggesting that pre- Crystal structures of related parainfluenza F glycoproteins have re- fusion and postfusion F may be antigenically distinct (11). To pre- vealed a large conformational change between the prefusion and vent viral entry, F-specific neutralizing antibodies presumably must postfusion states, suggesting that postfusion F antigens might not bind the prefusion conformation of F on the virion, before the viral efficiently elicit neutralizing antibodies. We have generated a ho- envelope fuses with a cellular membrane. Therefore, it might be mogeneous, stable, and reproducible postfusion RSV F immunogen expected that RSV F must be presented in the prefusion confor- that elicits high titers of neutralizing antibodies in immunized an- mation to elicit neutralizing antibodies efficiently. -

Preferred Product Characteristics of Monoclonal Antibodies for Passive
WHO/DRAFT-V0.1/MAR2020 1 2 3 Preferred Product Characteristics of Monoclonal Antibodies for Passive Immunization against Respiratory Syncytial Virus (RSV) 4 5 NOTE: 6 7 This draft has been prepared in consultation with the WHO Department of Immunization, Vaccines and Biologicals’ Technical Advisory Group for 8 RSV. 9 10 This document is being posted for the purpose of inviting comments and suggestions on the proposals contained therein, which will then be 11 considered by WHO's Product Development for Vaccines Advisory Committee (PDVAC) . 12 13 Written comments proposing modifications to this text must be received by 10 May 2020 and entered in the Comment Form (available 14 separately), and should be addressed to the Responsible Officer: Ms Erin Sparrow at [email protected]. 15 16 Background and purpose of WHO preferred product characteristics (PPCs) 17 This document describes World Health Organization (WHO) preferences for characteristics of monoclonal antibody (mAb) products used for 18 passive immunization against severe Respiratory Syncytial Virus (RSV) disease in infants. These preferences are shaped by the global unmet public 19 health need in priority disease areas for which WHO encourages the development of vaccines and other preventive interventions suitable for use 20 in low- and middle-income countries (LMICs). While many characteristics are the same as those preferred in high-income countries, there are 21 some characteristics that might be unique to LMIC settings.[1] 22 The primary audience for this document includes all involved in the development of new RSV mAbs intended for global use, that is contemplating 23 eventual WHO policy recommendation and prequalification. -

2006 Drug Trend Report
20 06 Vol 8 DRUGTREND REPORT 06MEDCODT_041906_COV_UG.indd 1 4/26/06 8:11:53 PM WE WOULD LIKE TO ACKNOWLEDGE THE FOLLOWING PEOPLE FOR THEIR OUTSTANDING CONTRIBUTIONS TO THE 2006 DRUG TREND REPORT: Ronald E. Aubert, Ph.D. Thomas Kellerhouse Beth Ann Bird, R.Ph., M.M. Mona Khalid, M.B.A. Mark Boyer JoAnn Krenitsky, M.P.H. Keith Bradbury, R.Ph., M.S. Wayne Levitt, M.H.H.A. Tracey Caldiero, M.B.A. Debra Ludgate Lon Castle, M.D. Debra A. Maldonato, M.S. Gina Catania Ken Malley, M.B.A. Kevin Cleary, Pharm.D., M.B.A. Barbara S. Menzel, M.P.H. Al DeCarlo Monique N. Naughton Michael Elefonte Glenn C. Nichols, M.P.A. Brad Epstein Elisabeth Ravazzolo, R.N., B.S.N. Robert S. Epstein, M.D., M.S. Steve Russek, R.Ph. Susan Faust Jodi Schreiber Jonah R. Gitlitz, M.B.A. Bill Schu WE WOULD LIKE TO ACKNOWLEDGE THE FOLLOWINGTracy Grunsfeld PEOPLE FOR THEIR Elizabeth Shee OUTSTANDING CONTRIBUTION TO THE MEDCOLydia 2004 Dixon DRUG Harden TREND REPORTJack: A. Smith, M.A. Glenn Herdling, M.S. Sue Steele Amy Aldighere; Berhanu Alemayehu, D.P.H.; Ron Aubert,Robert Hollis,Ph.D.; MarkM.B.A. Boyer; Keith Bradbury,Valerie Trainor R.Ph., M.S.; Jane Byron; Lon Castle, M.D.; Susan Dobek-Drew, Pharm.D.;Linda Huffer, Robert M.B.A. S. Epstein, M.D., M.S.;Robert Glenn R. Verbrugge, Herdling; Ph.D. Deepti Kehoe; Mona Khalid, M.B.A.; JoAnn Krenitsky,Susan M.P.H.; Jennings Barbara S. Menzel; Les StephenPaul, M.D., Wogen, M.S.; Alan M.H.A. -
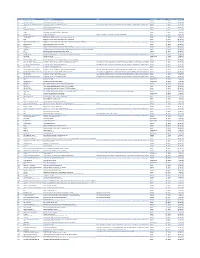
Cvx List.Pdf
CVX Code CVX Short Description Full Vaccine Name Note VaccineStatus internalID nonvaccine update_date 54 adenovirus, type 4 adenovirus vaccine, type 4, live, oral Inactive 1 False 28-May-10 55 adenovirus, type 7 adenovirus vaccine, type 7, live, oral Inactive 2 False 28-May-10 82 adenovirus, unspecified formulation adenovirus vaccine, unspecified formulation This CVX code allows reporting of a vaccination when formulation is unknown (for example, when reInactive 3 False 30-Sep-10 24 anthrax anthrax vaccine Active 4 False 11-Jun-19 19 BCG Bacillus Calmette-Guerin vaccine Active 5 False 28-May-10 27 botulinum antitoxin botulinum antitoxin Active 6 True 4-Sep-20 26 cholera, unspecified formulation cholera vaccine, unspecified formulation Inactive 7 False 17-Jun-16 29 CMVIG cytomegalovirus immune globulin, intravenous Active 8 True 4-Sep-20 56 dengue fever dengue fever vaccine Applies to dengue tetravalent product (e.g. DENGVAXIA) Active 9 False 24-Sep-19 12 diphtheria antitoxin diphtheria antitoxin Active 10 True 4-Sep-20 28 DT (pediatric) diphtheria and tetanus toxoids, adsorbed for pediatric use Active 11 False 28-May-10 20 DTaP diphtheria, tetanus toxoids and acellular pertussis vaccine Active 12 False 28-May-10 106 DTaP, 5 pertussis antigens diphtheria, tetanus toxoids and acellular pertussis vaccine, 5 pertussis antigens Active 13 False 28-May-10 107 DTaP, unspecified formulation diphtheria, tetanus toxoids and acellular pertussis vaccine, unspecified formulation This CVX code allows reporting of a vaccination when formulation is unknown