Epstein–Barr Virus Nuclear Antigen 3C Binds to BATF/IRF4 Or SPI1/IRF4 Composite Sites and Recruits Sin3a to Repress CDKN2A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Role of PU.1 and GATA-1 Transcription Factors During Normal and Leukemogenic Hematopoiesis
Leukemia (2010) 24, 1249–1257 & 2010 Macmillan Publishers Limited All rights reserved 0887-6924/10 www.nature.com/leu REVIEW The role of PU.1 and GATA-1 transcription factors during normal and leukemogenic hematopoiesis P Burda1, P Laslo2 and T Stopka1,3 1Department of Pathophysiology and Center of Experimental Hematology, First Faculty of Medicine, Charles University, Prague, Czech Republic; 2Section of Experimental Haematology, Leeds Institute of Molecular Medicine, University of Leads, St James’s University Hospital, Leeds, UK and 31st Department of Medicine-Hematology, General University Hospital, Prague, Czech Republic Hematopoiesis is coordinated by a complex regulatory network Additional domains include an N-terminal acidic domain and a of transcription factors and among them PU.1 (Spi1, Sfpi1) glutamine-rich domain, both involved in transcriptional activa- represents a key molecule. This review summarizes the tion, as well as a PEST domain involved in protein–protein indispensable requirement of PU.1 during hematopoietic cell fate decisions and how the function of PU.1 can be modulated interactions. PU.1 protein can be modified post-translationally by protein–protein interactions with additional factors. The by phosporylation at serines 41 (N-terminal acidic domain) and mutual negative regulation between PU.1 and GATA-1 is 142 and 148 (PEST domain), which results in augmented detailed within the context of normal and leukemogenic activity. hematopoiesis and the concept of ‘differentiation therapy’ to The PU.1 protein can physically interact with a variety of restore normal cellular differentiation of leukemic cells is regulatory factors including (i) general transcription factors discussed. Leukemia (2010) 24, 1249–1257; doi:10.1038/leu.2010.104; (TFIID, TBP), (ii) early hematopoietic transcription factors published online 3 June 2010 (GATA-2 and Runx-1), (iii) erythroid factor (GATA-1) and (iv) Keywords: PU.1; leukemia differentiation; GATA-1; chromatin; non-erythroid factors (C/EBPa, C/EBPb, IRF4/8 and c-Jun). -

Activated Peripheral-Blood-Derived Mononuclear Cells
Transcription factor expression in lipopolysaccharide- activated peripheral-blood-derived mononuclear cells Jared C. Roach*†, Kelly D. Smith*‡, Katie L. Strobe*, Stephanie M. Nissen*, Christian D. Haudenschild§, Daixing Zhou§, Thomas J. Vasicek¶, G. A. Heldʈ, Gustavo A. Stolovitzkyʈ, Leroy E. Hood*†, and Alan Aderem* *Institute for Systems Biology, 1441 North 34th Street, Seattle, WA 98103; ‡Department of Pathology, University of Washington, Seattle, WA 98195; §Illumina, 25861 Industrial Boulevard, Hayward, CA 94545; ¶Medtronic, 710 Medtronic Parkway, Minneapolis, MN 55432; and ʈIBM Computational Biology Center, P.O. Box 218, Yorktown Heights, NY 10598 Contributed by Leroy E. Hood, August 21, 2007 (sent for review January 7, 2007) Transcription factors play a key role in integrating and modulating system. In this model system, we activated peripheral-blood-derived biological information. In this study, we comprehensively measured mononuclear cells, which can be loosely termed ‘‘macrophages,’’ the changing abundances of mRNAs over a time course of activation with lipopolysaccharide (LPS). We focused on the precise mea- of human peripheral-blood-derived mononuclear cells (‘‘macro- surement of mRNA concentrations. There is currently no high- phages’’) with lipopolysaccharide. Global and dynamic analysis of throughput technology that can precisely and sensitively measure all transcription factors in response to a physiological stimulus has yet to mRNAs in a system, although such technologies are likely to be be achieved in a human system, and our efforts significantly available in the near future. To demonstrate the potential utility of advanced this goal. We used multiple global high-throughput tech- such technologies, and to motivate their development and encour- nologies for measuring mRNA levels, including massively parallel age their use, we produced data from a combination of two distinct signature sequencing and GeneChip microarrays. -

The Unique Transcriptional Response Produced by Concurrent Estrogen
Need et al. BMC Cancer (2015) 15:791 DOI 10.1186/s12885-015-1819-3 RESEARCH ARTICLE Open Access The unique transcriptional response produced by concurrent estrogen and progesterone treatment in breast cancer cells results in upregulation of growth factor pathways and switching from a Luminal A to a Basal-like subtype Eleanor F. Need1*,LukeA.Selth2,3,AndrewP.Trotta1,4,DamienA.Leach1,LaurenGiorgio1, Melissa A. O’Loughlin1, Eric Smith5, Peter G. Gill6,WendyV.Ingman7,8, J. Dinny Graham9 and Grant Buchanan1,3 Abstract Background: In breast cancer, progesterone receptor (PR) positivity or abundance is positively associated with survival and treatment response. It was initially believed that PR was a useful diagnostic marker of estrogen receptor activity, but increasingly PR has been recognised to play an important biological role in breast homeostasis, carcinogenesis and metastasis. Although PR expression is almost exclusively observed in estrogen receptor positive tumors, few studies have investigated the cellular mechanisms of PR action in the context of ongoing estrogen signalling. Methods: In this study, we contrast PR function in estrogen pretreated ZR-75-1 breast cancer cells with vehicle treated ZR-75-1 and T-47D breast cancer cells using expression microarrays and chromatin immunoprecipitation-sequencing. Results: Estrogen cotreatment caused a dramatic increase in the number of genes regulated by progesterone in ZR-75-1 cells. In T-47D cells that have naturally high levels of PR, estrogen and progesterone cotreatment resulted in a reduction in the number of regulated genes in comparison to treatment with either hormone alone. At a genome level, estrogen pretreatment of ZR-75-1 cells led to a 10-fold increase in the number of PR DNA binding sites detected using ChIP-sequencing. -

Discovery of Progenitor Cell Signatures by Time-Series Synexpression Analysis During Drosophila Embryonic Cell Immortalization
Correction DEVELOPMENTAL BIOLOGY Correction for “Discovery of progenitor cell signatures by time- series synexpression analysis during Drosophila embryonic cell immortalization,” by Mary-Lee Dequéant, Delphine Fagegaltier, Yanhui Hu, Kerstin Spirohn, Amanda Simcox, Gregory J. Hannon, and Norbert Perrimon, which appeared in issue 42, October 20, 2015, of Proc Natl Acad Sci USA (112:12974–12979; first published October 5, 2015; 10.1073/pnas.1517729112). The authors note that Delphine Fagegaltier should be credited for designing research and performing research. The authors also note that Delphine Fagegaltier, Amanda Simcox, and Gregory J. Hannon should be credited for contributing to the writing of the paper. The corrected author contributions footnote appears below. Author contributions: M.-L.D., D.F., A.S., G.J.H., and N.P. designed research; M.-L.D., D.F., K.S., and A.S. performed research; M.-L.D., D.F., and Y.H. analyzed data; and M.-L.D. and N.P. wrote the paper with contributions from D.F., A.S., and G.J.H. www.pnas.org/cgi/doi/10.1073/pnas.1520482112 E6408 | PNAS | November 17, 2015 | vol. 112 | no. 46 www.pnas.org Downloaded by guest on September 25, 2021 Discovery of progenitor cell signatures by time-series synexpression analysis during Drosophila embryonic cell immortalization Mary-Lee Dequéanta,1, Delphine Fagegaltierb, Yanhui Hua, Kerstin Spirohna, Amanda Simcoxc, Gregory J. Hannond, and Norbert Perrimona,e,1 aDepartment of Genetics, Harvard Medical School, Boston, MA 02115, bCold Spring Harbor Laboratories, Cold Spring Harbor, NY 11724; cDepartment of Molecular Genetics, The Ohio State University, Columbus, OH 43210; dHoward Hughes Medical Institute, Cold Spring Harbor Laboratories, Cold Spring Harbor, NY 11724; and eHoward Hughes Medical Institute, Harvard Medical School, Boston, MA 02115 Contributed by Norbert Perrimon, September 10, 2015 (sent for review May 18, 2015; reviewed by Peter Cherbas, Gary Karpen, and Renato Paro) The use of time series profiling to identify groups of functionally population contributing to adult muscles (4–7). -

HSF1) During Macrophage Differentiation of Monocytes
Leukemia (2014) 28, 1676–1686 & 2014 Macmillan Publishers Limited All rights reserved 0887-6924/14 www.nature.com/leu ORIGINAL ARTICLE Dual regulation of SPI1/PU.1 transcription factor by heat shock factor 1 (HSF1) during macrophage differentiation of monocytes G Jego1,2, D Lanneau1,2, A De Thonel1,2, K Berthenet1,2, A Hazoume´ 1,2, N Droin3,4, A Hamman1,2, F Girodon1,2, P-S Bellaye1,2, G Wettstein1,2, A Jacquel1,2,5, L Duplomb6,7, A Le Moue¨l8,9, C Papanayotou10, E Christians11, P Bonniaud1,2, V Lallemand-Mezger8,9, E Solary3,4 and C Garrido1,2,12 In addition to their cytoprotective role in stressful conditions, heat shock proteins (HSPs) are involved in specific differentiation pathways, for example, we have identified a role for HSP90 in macrophage differentiation of human peripheral blood monocytes that are exposed to macrophage colony-stimulating factor (M-CSF). Here, we show that deletion of the main transcription factor involved in heat shock gene regulation, heat shock factor 1 (HSF1), affects M-CSF-driven differentiation of mouse bone marrow cells. HSF1 transiently accumulates in the nucleus of human monocytes undergoing macrophage differentiation, including M-CSF- treated peripheral blood monocytes and phorbol ester-treated THP1 cells. We demonstrate that HSF1 has a dual effect on SPI1/ PU.1, a transcription factor essential for macrophage differentiation and whose deregulation can lead to the development of leukemias and lymphomas. Firstly, HSF1 regulates SPI1/PU.1 gene expression through its binding to a heat shock element within the intron 2 of this gene. -

Inhibition of Mitochondrial Complex II in Neuronal Cells Triggers Unique
www.nature.com/scientificreports OPEN Inhibition of mitochondrial complex II in neuronal cells triggers unique pathways culminating in autophagy with implications for neurodegeneration Sathyanarayanan Ranganayaki1, Neema Jamshidi2, Mohamad Aiyaz3, Santhosh‑Kumar Rashmi4, Narayanappa Gayathri4, Pulleri Kandi Harsha5, Balasundaram Padmanabhan6 & Muchukunte Mukunda Srinivas Bharath7* Mitochondrial dysfunction and neurodegeneration underlie movement disorders such as Parkinson’s disease, Huntington’s disease and Manganism among others. As a corollary, inhibition of mitochondrial complex I (CI) and complex II (CII) by toxins 1‑methyl‑4‑phenylpyridinium (MPP+) and 3‑nitropropionic acid (3‑NPA) respectively, induced degenerative changes noted in such neurodegenerative diseases. We aimed to unravel the down‑stream pathways associated with CII inhibition and compared with CI inhibition and the Manganese (Mn) neurotoxicity. Genome‑wide transcriptomics of N27 neuronal cells exposed to 3‑NPA, compared with MPP+ and Mn revealed varied transcriptomic profle. Along with mitochondrial and synaptic pathways, Autophagy was the predominant pathway diferentially regulated in the 3‑NPA model with implications for neuronal survival. This pathway was unique to 3‑NPA, as substantiated by in silico modelling of the three toxins. Morphological and biochemical validation of autophagy markers in the cell model of 3‑NPA revealed incomplete autophagy mediated by mechanistic Target of Rapamycin Complex 2 (mTORC2) pathway. Interestingly, Brain Derived Neurotrophic Factor -

Appendix 2. Significantly Differentially Regulated Genes in Term Compared with Second Trimester Amniotic Fluid Supernatant
Appendix 2. Significantly Differentially Regulated Genes in Term Compared With Second Trimester Amniotic Fluid Supernatant Fold Change in term vs second trimester Amniotic Affymetrix Duplicate Fluid Probe ID probes Symbol Entrez Gene Name 1019.9 217059_at D MUC7 mucin 7, secreted 424.5 211735_x_at D SFTPC surfactant protein C 416.2 206835_at STATH statherin 363.4 214387_x_at D SFTPC surfactant protein C 295.5 205982_x_at D SFTPC surfactant protein C 288.7 1553454_at RPTN repetin solute carrier family 34 (sodium 251.3 204124_at SLC34A2 phosphate), member 2 238.9 206786_at HTN3 histatin 3 161.5 220191_at GKN1 gastrokine 1 152.7 223678_s_at D SFTPA2 surfactant protein A2 130.9 207430_s_at D MSMB microseminoprotein, beta- 99.0 214199_at SFTPD surfactant protein D major histocompatibility complex, class II, 96.5 210982_s_at D HLA-DRA DR alpha 96.5 221133_s_at D CLDN18 claudin 18 94.4 238222_at GKN2 gastrokine 2 93.7 1557961_s_at D LOC100127983 uncharacterized LOC100127983 93.1 229584_at LRRK2 leucine-rich repeat kinase 2 HOXD cluster antisense RNA 1 (non- 88.6 242042_s_at D HOXD-AS1 protein coding) 86.0 205569_at LAMP3 lysosomal-associated membrane protein 3 85.4 232698_at BPIFB2 BPI fold containing family B, member 2 84.4 205979_at SCGB2A1 secretoglobin, family 2A, member 1 84.3 230469_at RTKN2 rhotekin 2 82.2 204130_at HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 81.9 222242_s_at KLK5 kallikrein-related peptidase 5 77.0 237281_at AKAP14 A kinase (PRKA) anchor protein 14 76.7 1553602_at MUCL1 mucin-like 1 76.3 216359_at D MUC7 mucin 7, -

Functional Characterization of TBR1 Variants in Neurodevelopmental Disorder Received: 14 May 2018 Joery Den Hoed1, Elliot Sollis1, Hanka Venselaar2, Sara B
www.nature.com/scientificreports OPEN Functional characterization of TBR1 variants in neurodevelopmental disorder Received: 14 May 2018 Joery den Hoed1, Elliot Sollis1, Hanka Venselaar2, Sara B. Estruch1, Pelagia Deriziotis1 & Accepted: 31 August 2018 Simon E. Fisher 1,3 Published: xx xx xxxx Recurrent de novo variants in the TBR1 transcription factor are implicated in the etiology of sporadic autism spectrum disorders (ASD). Disruptions include missense variants located in the T-box DNA- binding domain and previous work has demonstrated that they disrupt TBR1 protein function. Recent screens of thousands of simplex families with sporadic ASD cases uncovered additional T-box variants in TBR1 but their etiological relevance is unclear. We performed detailed functional analyses of de novo missense TBR1 variants found in the T-box of ASD cases, assessing many aspects of protein function, including subcellular localization, transcriptional activity and protein-interactions. Only two of the three tested variants severely disrupted TBR1 protein function, despite in silico predictions that all would be deleterious. Furthermore, we characterized a putative interaction with BCL11A, a transcription factor that was recently implicated in a neurodevelopmental syndrome involving developmental delay and language defcits. Our fndings enhance understanding of molecular functions of TBR1, as well as highlighting the importance of functional testing of variants that emerge from next-generation sequencing, to decipher their contributions to neurodevelopmental disorders like ASD. TBR1 (T-box brain, 1; OMIM *604616) encodes a neuron-specifc transcription factor of the T-box family1. Te TBR1 protein is highly expressed in the deep layers of the cortex, where it is involved in diferentiation of subsets of projection neurons2–4. -

Reduction of BCL11A in Hematopoietic Stem Cells Through
Science Bulletin 64 (2019) 1562–1564 Contents lists available at ScienceDirect Science Bulletin journal homepage: www.elsevier.com/locate/scib Research Highlight Reduction of BCL11A in hematopoietic stem cells through gene editing: new strategy to ameliorate the severe b-globin disorders sickle cell disease ⇑ Weiqi Hong, Mengyuan Huang, Yuquan Wei, Xiawei Wei Laboratory of Aging Research and Cancer Drug Target, State Key Laboratory of Biotherapy, National Clinical Research Center for Geriatrics, West China Hospital, Sichuan University, Chengdu 610041, China Site-specific gene editing is of great importance in precise Sickle-cell anemia is a prototypical monogenic disorder caused medicine. Two conventional genome editing methods, Zine finger by mutation of b-globin subunit. It is a promising therapy strategy nucleases (ZFNs) and transcription activator-like effector nucleases to induct fetal hemoglobin (HbF, a2c2) by re-expressing the paral- (TALENs), are based on protein-DNA recognition, with tedious ogous c-globin genes (HBG1/2) for severe b-globin disorders sickle work in constructing target protein [1,2]. Developed from immune cell disease (SCD) and b-thalassemia [9]. Researches in the past response of bacteria, CRISPR/Cas9 has been widely investigated as have shown that the core of the +58 erythroid enhancer of BCL11A a promising tool for therapeutic genome editing in clinical settings was crucial for repression of HBF in adult stage erythroid. Wu et al nowadays [3,4]. This system succeeds in gene deletion, insertion found that chemically modified synthetic sgRNAs (MS-sgRNAs) and frameshift mutations with higher efficiency, less cost, was more efficient than in vitro transcribed sgRNAs. Targeting improved flexibility and simplified designing process [5]. -
Type of the Paper (Article
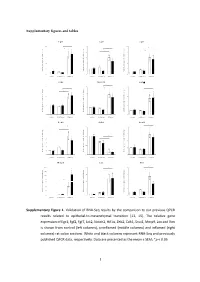
Supplementary figures and tables E g r 1 F g f2 F g f7 1 0 * 5 1 0 * * e e e * g g g * n n n * a a a 8 4 * 8 h h h * c c c d d d * l l l o o o * f f f * n n n o o o 6 3 6 i i i s s s s s s e e e r r r p p p x x x e e e 4 2 4 e e e n n n e e e g g g e e e v v v i i i t t t 2 1 2 a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d J a k 2 N o tc h 2 H if1 * 3 4 6 * * * e e e g g g n n n a a * * a * h h * h c c c 3 * d d * d l l l * o o o f f 2 f 4 n n n o o o i i i s s s s s s e e e r r 2 r p p p x x x e e e e e e n n n e e 1 e 2 g g g e e 1 e v v v i i i t t t a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Z e b 2 C d h 1 S n a i1 * * 7 1 .5 4 * * e e e g g g 6 n n n * a a a * h h h c c c 3 * d d d l l l 5 o o o f f f 1 .0 * n n n * o o o i i i 4 * s s s s s s e e e r r r 2 p p p x x x 3 e e e e e e n n n e e e 0 .5 g g g 2 e e e 1 v v v i i i t t t a a a * l l l e e e 1 * R R R 0 0 .0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d M m p 9 L o x V im 2 0 0 2 0 8 * * * e e e * g g g 1 5 0 * n n n * a a a * h h h * c c c 1 5 * 6 d d d l l l 1 0 0 o o o f f f n n n o o o i i i 5 0 s s s s s s * e e e r r r 1 0 4 3 0 p p p * x x x e e e * e e e n n n e e e 2 0 g g g e e e 5 2 v v v i i i t t t a a a l l l 1 0 e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Supplementary Figure 1. -

Transcriptome Alterations of Vascular Smooth Muscle Cells in Aortic Wall of Myocardial Infarction Patients
This document is downloaded from DR‑NTU (https://dr.ntu.edu.sg) Nanyang Technological University, Singapore. Transcriptome alterations of vascular smooth muscle cells in aortic wall of myocardial infarction patients Wongsurawat, Thidathip; Woo, Chin Cheng; Giannakakis, Antonis; Lin, Xiao Yun; Cheow, Esther Sok Hwee; Lee, Chuen Neng; Richards, Mark; Sze, Siu Kwan; Nookaew, Intawat; Sorokin, Vitaly; Kuznetsov, Vladimir Andreevich 2018 Wongsurawat, T., Woo, C. C., Giannakakis, A., Lin, X. Y., Cheow, E. S. H., Lee, C. N., et al. (2018). Transcriptome alterations of vascular smooth muscle cells in aortic wall of myocardial infarction patients. Data in Brief, 17, 1112‑1135. https://hdl.handle.net/10356/85590 https://doi.org/10.1016/j.dib.2018.01.108 © 2018 The Authors. Published by Elsevier Inc. This is an open access article under the CC BY license (http://creativecommons.org/licenses/by/4.0/). Downloaded on 09 Oct 2021 06:21:01 SGT Data in Brief 17 (2018) 1112–1135 Contents lists available at ScienceDirect Data in Brief journal homepage: www.elsevier.com/locate/dib Data Article Transcriptome alterations of vascular smooth muscle cells in aortic wall of myocardial infarction patients Thidathip Wongsurawat a,b, Chin Cheng Woo c, Antonis Giannakakis a, Xiao Yun Lin d, Esther Sok Hwee Cheow e, Chuen Neng Lee c,d, Mark Richards f,g, Siu Kwan Sze e, Intawat Nookaew b, Vladimir A. Kuznetsov a,h, Vitaly Sorokin c,d,⁎ a Department of Genome and Gene Expression Data Analysis, Bioinformatics Institute, Agency for Science, Technology and Research (A*STAR), -

Functional Annotation of Human Long Noncoding Rnas Using Chromatin
bioRxiv preprint doi: https://doi.org/10.1101/2021.01.13.426305; this version posted January 14, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Funconal annotaon of human long noncoding RNAs using chroman conformaon data Saumya Agrawal1, Tanvir Alam2, Masaru Koido1,3, Ivan V. Kulakovskiy4,5, Jessica Severin1, Imad ABugessaisa1, Andrey Buyan5,6, Josee Dos&e7, Masayoshi Itoh1,8, Naoto Kondo9, Yunjing Li10, Mickaël Mendez11, Jordan A. Ramilowski1,12, Ken Yagi1, Kayoko Yasuzawa1, CHi Wai Yip1, Yasushi Okazaki1, MicHael M. Ho9man11,13,14,15, Lisa Strug10, CHung CHau Hon1, CHikashi Terao1, Takeya Kasukawa1, Vsevolod J. Makeev4,16, Jay W. SHin1, Piero Carninci1, MicHiel JL de Hoon1 1RIKEN Center for Integra&ve Medical Sciences, YokoHama, Japan. 2College of Science and Engineering, Hamad Bin KHalifa University, DoHa, Qatar. 3Ins&tute of Medical Science, THe University of Tokyo, Tokyo, Japan. 4Vavilov Ins&tute of General Gene&cs, Russian Academy of Sciences, Moscow, Russia. 5Ins&tute of Protein ResearcH, Russian Academy of Sciences, PusHcHino, Russia. 6Faculty of Bioengineering and Bioinforma&cs, Lomonosov Moscow State University, Moscow, Russia. 7Department of BiocHemistry, Rosalind and Morris Goodman Cancer ResearcH Center, McGill University, Montréal, QuéBec, Canada. 8RIKEN Preven&ve Medicine and Diagnosis Innova&on Program, Wako, Japan. 9RIKEN Center for Life Science TecHnologies, YokoHama, Japan. 10Division of Biosta&s&cs, Dalla Lana ScHool of PuBlic HealtH, University of Toronto, Toronto, Ontario, Canada. 11Department of Computer Science, University of Toronto, Toronto, Ontario, Canada. 12Advanced Medical ResearcH Center, YokoHama City University, YokoHama, Japan.