Logical Modeling of Lymphoid and Myeloid Cell Specification and Transdifferentiation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Role of PU.1 and GATA-1 Transcription Factors During Normal and Leukemogenic Hematopoiesis
Leukemia (2010) 24, 1249–1257 & 2010 Macmillan Publishers Limited All rights reserved 0887-6924/10 www.nature.com/leu REVIEW The role of PU.1 and GATA-1 transcription factors during normal and leukemogenic hematopoiesis P Burda1, P Laslo2 and T Stopka1,3 1Department of Pathophysiology and Center of Experimental Hematology, First Faculty of Medicine, Charles University, Prague, Czech Republic; 2Section of Experimental Haematology, Leeds Institute of Molecular Medicine, University of Leads, St James’s University Hospital, Leeds, UK and 31st Department of Medicine-Hematology, General University Hospital, Prague, Czech Republic Hematopoiesis is coordinated by a complex regulatory network Additional domains include an N-terminal acidic domain and a of transcription factors and among them PU.1 (Spi1, Sfpi1) glutamine-rich domain, both involved in transcriptional activa- represents a key molecule. This review summarizes the tion, as well as a PEST domain involved in protein–protein indispensable requirement of PU.1 during hematopoietic cell fate decisions and how the function of PU.1 can be modulated interactions. PU.1 protein can be modified post-translationally by protein–protein interactions with additional factors. The by phosporylation at serines 41 (N-terminal acidic domain) and mutual negative regulation between PU.1 and GATA-1 is 142 and 148 (PEST domain), which results in augmented detailed within the context of normal and leukemogenic activity. hematopoiesis and the concept of ‘differentiation therapy’ to The PU.1 protein can physically interact with a variety of restore normal cellular differentiation of leukemic cells is regulatory factors including (i) general transcription factors discussed. Leukemia (2010) 24, 1249–1257; doi:10.1038/leu.2010.104; (TFIID, TBP), (ii) early hematopoietic transcription factors published online 3 June 2010 (GATA-2 and Runx-1), (iii) erythroid factor (GATA-1) and (iv) Keywords: PU.1; leukemia differentiation; GATA-1; chromatin; non-erythroid factors (C/EBPa, C/EBPb, IRF4/8 and c-Jun). -

Activated Peripheral-Blood-Derived Mononuclear Cells
Transcription factor expression in lipopolysaccharide- activated peripheral-blood-derived mononuclear cells Jared C. Roach*†, Kelly D. Smith*‡, Katie L. Strobe*, Stephanie M. Nissen*, Christian D. Haudenschild§, Daixing Zhou§, Thomas J. Vasicek¶, G. A. Heldʈ, Gustavo A. Stolovitzkyʈ, Leroy E. Hood*†, and Alan Aderem* *Institute for Systems Biology, 1441 North 34th Street, Seattle, WA 98103; ‡Department of Pathology, University of Washington, Seattle, WA 98195; §Illumina, 25861 Industrial Boulevard, Hayward, CA 94545; ¶Medtronic, 710 Medtronic Parkway, Minneapolis, MN 55432; and ʈIBM Computational Biology Center, P.O. Box 218, Yorktown Heights, NY 10598 Contributed by Leroy E. Hood, August 21, 2007 (sent for review January 7, 2007) Transcription factors play a key role in integrating and modulating system. In this model system, we activated peripheral-blood-derived biological information. In this study, we comprehensively measured mononuclear cells, which can be loosely termed ‘‘macrophages,’’ the changing abundances of mRNAs over a time course of activation with lipopolysaccharide (LPS). We focused on the precise mea- of human peripheral-blood-derived mononuclear cells (‘‘macro- surement of mRNA concentrations. There is currently no high- phages’’) with lipopolysaccharide. Global and dynamic analysis of throughput technology that can precisely and sensitively measure all transcription factors in response to a physiological stimulus has yet to mRNAs in a system, although such technologies are likely to be be achieved in a human system, and our efforts significantly available in the near future. To demonstrate the potential utility of advanced this goal. We used multiple global high-throughput tech- such technologies, and to motivate their development and encour- nologies for measuring mRNA levels, including massively parallel age their use, we produced data from a combination of two distinct signature sequencing and GeneChip microarrays. -

The REST/NRSF Pathway As a Central Mechanism in CNS Dysfunction
The REST/NRSF Pathway as a Central Mechanism in CNS Dysfunction Thesis submitted in accordance with the requirements of the University of Liverpool for the degree of Doctor in Philosophy by Alix Warburton April 2015 Disclaimer The data in this thesis is a result of my own work. The material collected for this thesis has not been presented, nor is currently being presented, either wholly or in part for any other degree or other qualification. All of the research, unless otherwise stated, was performed in the Department of Physiology and Department of Pharmacology, Institute of Translational Medicine, University of Liverpool. All other parties involved in the research presented here, and the nature of their contribution, are listed in the Acknowledgements section of this thesis. i Acknowledgements First and foremost, I would like to express my upmost gratitude to my primary and secondary supervisors Professor John Quinn (a.k.a Prof. Quinny) and Dr Jill Bubb for all of their support, guidance, wisdom (thank you Jill) and encouragement throughout my PhD; I could not have wished for a better pair. I am also extremely grateful to the BBSRC for funding my PhD project. I would also like to extend my thanks to Dr Graeme Sills for providing samples and assistance with my work on the SANAD epilepsy project, Dr Fabio Miyajima for offering his knowledge and knowhow on many occasions, Dr Gerome Breen for being a bioinformatics wizard and providing support on several projects, Dr Minyan Wang’s lab for their help and hospitality during my 3 month visit to Xi'an Jiaotong-Liverpool University, Dr Roshan Koron for assisting with the breast cancer study, Dr Chris Murgatroyd for his invaluable advice on ChIP and Professor Dan Rujescu’s lab for providing clinical samples and support with statistical analyses on the schizophrenia project. -

The Unique Transcriptional Response Produced by Concurrent Estrogen
Need et al. BMC Cancer (2015) 15:791 DOI 10.1186/s12885-015-1819-3 RESEARCH ARTICLE Open Access The unique transcriptional response produced by concurrent estrogen and progesterone treatment in breast cancer cells results in upregulation of growth factor pathways and switching from a Luminal A to a Basal-like subtype Eleanor F. Need1*,LukeA.Selth2,3,AndrewP.Trotta1,4,DamienA.Leach1,LaurenGiorgio1, Melissa A. O’Loughlin1, Eric Smith5, Peter G. Gill6,WendyV.Ingman7,8, J. Dinny Graham9 and Grant Buchanan1,3 Abstract Background: In breast cancer, progesterone receptor (PR) positivity or abundance is positively associated with survival and treatment response. It was initially believed that PR was a useful diagnostic marker of estrogen receptor activity, but increasingly PR has been recognised to play an important biological role in breast homeostasis, carcinogenesis and metastasis. Although PR expression is almost exclusively observed in estrogen receptor positive tumors, few studies have investigated the cellular mechanisms of PR action in the context of ongoing estrogen signalling. Methods: In this study, we contrast PR function in estrogen pretreated ZR-75-1 breast cancer cells with vehicle treated ZR-75-1 and T-47D breast cancer cells using expression microarrays and chromatin immunoprecipitation-sequencing. Results: Estrogen cotreatment caused a dramatic increase in the number of genes regulated by progesterone in ZR-75-1 cells. In T-47D cells that have naturally high levels of PR, estrogen and progesterone cotreatment resulted in a reduction in the number of regulated genes in comparison to treatment with either hormone alone. At a genome level, estrogen pretreatment of ZR-75-1 cells led to a 10-fold increase in the number of PR DNA binding sites detected using ChIP-sequencing. -

Mediator of DNA Damage Checkpoint 1 (MDC1) Is a Novel Estrogen Receptor Co-Regulator in Invasive 6 Lobular Carcinoma of the Breast 7 8 Evelyn K
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.16.423142; this version posted December 16, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 1 Running Title: MDC1 co-regulates ER in ILC 2 3 Research article 4 5 Mediator of DNA damage checkpoint 1 (MDC1) is a novel estrogen receptor co-regulator in invasive 6 lobular carcinoma of the breast 7 8 Evelyn K. Bordeaux1+, Joseph L. Sottnik1+, Sanjana Mehrotra1, Sarah E. Ferrara2, Andrew E. Goodspeed2,3, James 9 C. Costello2,3, Matthew J. Sikora1 10 11 +EKB and JLS contributed equally to this project. 12 13 Affiliations 14 1Dept. of Pathology, University of Colorado Anschutz Medical Campus 15 2Biostatistics and Bioinformatics Shared Resource, University of Colorado Comprehensive Cancer Center 16 3Dept. of Pharmacology, University of Colorado Anschutz Medical Campus 17 18 Corresponding author 19 Matthew J. Sikora, PhD.; Mail Stop 8104, Research Complex 1 South, Room 5117, 12801 E. 17th Ave.; Aurora, 20 CO 80045. Tel: (303)724-4301; Fax: (303)724-3712; email: [email protected]. Twitter: 21 @mjsikora 22 23 Authors' contributions 24 MJS conceived of the project. MJS, EKB, and JLS designed and performed experiments. JLS developed models 25 for the project. EKB, JLS, SM, and AEG contributed to data analysis and interpretation. SEF, AEG, and JCC 26 developed and performed informatics analyses. MJS wrote the draft manuscript; all authors read and revised the 27 manuscript and have read and approved of this version of the manuscript. -

Intrinsic Specificity of DNA Binding and Function of Class II Bhlh
INTRINSIC SPECIFICITY OF BINDING AND REGULATORY FUNCTION OF CLASS II BHLH TRANSCRIPTION FACTORS APPROVED BY SUPERVISORY COMMITTEE Jane E. Johnson Ph.D. Helmut Kramer Ph.D. Genevieve Konopka Ph.D. Raymond MacDonald Ph.D. INTRINSIC SPECIFICITY OF BINDING AND REGULATORY FUNCTION OF CLASS II BHLH TRANSCRIPTION FACTORS by BRADFORD HARRIS CASEY DISSERTATION Presented to the Faculty of the Graduate School of Biomedical Sciences The University of Texas Southwestern Medical Center at Dallas In Partial Fulfillment of the Requirements For the Degree of DOCTOR OF PHILOSOPHY The University of Texas Southwestern Medical Center at Dallas Dallas, Texas December, 2016 DEDICATION This work is dedicated to my family, who have taught me pursue truth in all forms. To my grandparents for inspiring my curiosity, my parents for teaching me the value of a life in the service of others, my sisters for reminding me of the importance of patience, and to Rachel, who is both “the beautiful one”, and “the smart one”, and insists that I am clever and beautiful, too. Copyright by Bradford Harris Casey, 2016 All Rights Reserved INTRINSIC SPECIFICITY OF BINDING AND REGULATORY FUNCTION OF CLASS II BHLH TRANSCRIPTION FACTORS Publication No. Bradford Harris Casey The University of Texas Southwestern Medical Center at Dallas, 2016 Jane E. Johnson, Ph.D. PREFACE Embryonic development begins with a single cell, and gives rise to the many diverse cells which comprise the complex structures of the adult animal. Distinct cell fates require precise regulation to develop and maintain their functional characteristics. Transcription factors provide a mechanism to select tissue-specific programs of gene expression from the shared genome. -

Accompanies CD8 T Cell Effector Function Global DNA Methylation
Global DNA Methylation Remodeling Accompanies CD8 T Cell Effector Function Christopher D. Scharer, Benjamin G. Barwick, Benjamin A. Youngblood, Rafi Ahmed and Jeremy M. Boss This information is current as of October 1, 2021. J Immunol 2013; 191:3419-3429; Prepublished online 16 August 2013; doi: 10.4049/jimmunol.1301395 http://www.jimmunol.org/content/191/6/3419 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130139 Material 5.DC1 References This article cites 81 articles, 25 of which you can access for free at: http://www.jimmunol.org/content/191/6/3419.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on October 1, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Global DNA Methylation Remodeling Accompanies CD8 T Cell Effector Function Christopher D. Scharer,* Benjamin G. Barwick,* Benjamin A. Youngblood,*,† Rafi Ahmed,*,† and Jeremy M. -

HSF1) During Macrophage Differentiation of Monocytes
Leukemia (2014) 28, 1676–1686 & 2014 Macmillan Publishers Limited All rights reserved 0887-6924/14 www.nature.com/leu ORIGINAL ARTICLE Dual regulation of SPI1/PU.1 transcription factor by heat shock factor 1 (HSF1) during macrophage differentiation of monocytes G Jego1,2, D Lanneau1,2, A De Thonel1,2, K Berthenet1,2, A Hazoume´ 1,2, N Droin3,4, A Hamman1,2, F Girodon1,2, P-S Bellaye1,2, G Wettstein1,2, A Jacquel1,2,5, L Duplomb6,7, A Le Moue¨l8,9, C Papanayotou10, E Christians11, P Bonniaud1,2, V Lallemand-Mezger8,9, E Solary3,4 and C Garrido1,2,12 In addition to their cytoprotective role in stressful conditions, heat shock proteins (HSPs) are involved in specific differentiation pathways, for example, we have identified a role for HSP90 in macrophage differentiation of human peripheral blood monocytes that are exposed to macrophage colony-stimulating factor (M-CSF). Here, we show that deletion of the main transcription factor involved in heat shock gene regulation, heat shock factor 1 (HSF1), affects M-CSF-driven differentiation of mouse bone marrow cells. HSF1 transiently accumulates in the nucleus of human monocytes undergoing macrophage differentiation, including M-CSF- treated peripheral blood monocytes and phorbol ester-treated THP1 cells. We demonstrate that HSF1 has a dual effect on SPI1/ PU.1, a transcription factor essential for macrophage differentiation and whose deregulation can lead to the development of leukemias and lymphomas. Firstly, HSF1 regulates SPI1/PU.1 gene expression through its binding to a heat shock element within the intron 2 of this gene. -

Early B Cell Factor 1 Regulates B Cell Gene Networks by Activation, Repression, and Transcription- Independent Poising of Chromatin
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Immunity Resource Early B Cell Factor 1 Regulates B Cell Gene Networks by Activation, Repression, and Transcription- Independent Poising of Chromatin Thomas Treiber,1,3 Elizabeth M. Mandel,1,3 Sebastian Pott,2,3 Ildiko Gyo¨ ry,1,3 Sonja Firner,1 Edison T. Liu,2 and Rudolf Grosschedl1,* 1Max Planck Institute of Immunobiology, Department of Cellular and Molecular Immunology, Stuebeweg 51, 79108 Freiburg, Germany 2Genome Institute of Singapore, Cancer Biology and Pharmacology, 60 Biopolis Street, 138672 Singapore 3These authors contributed equally to this work *Correspondence: [email protected] DOI 10.1016/j.immuni.2010.04.013 SUMMARY (reviewed in Hardy et al., 2007; Murre, 2009). Pre-B cells express the pre-B cell receptor (pre-BCR) and further differentiate into The transcription factor early B cell factor-1 (Ebf1) is immature B cells that have undergone Ig light chain gene rear- a key determinant of B lineage specification and rangement and migrate from the bone marrow to the spleen. differentiation. To gain insight into the molecular Each of these B cell differentiation steps is dependent on basis of Ebf1 function in early-stage B cells, we the coordinated expression of cell-type-specific transcription combined a genome-wide ChIP sequencing analysis factors and activities of signaling pathways (reviewed in Mandel with gain- and loss-of-function transcriptome anal- and Grosschedl, 2010). Genetic ablation and complementation studies have demonstrated key roles for transcription factors yses. Among 565 genes that are occupied and tran- such as Ikaros, Pu.1, E2A, early B cell factor-1 (Ebf1), and scriptionally regulated by Ebf1, we identified large Pax5 (reviewed in Busslinger, 2004; Hagman and Lukin, 2006; sets involved in (pre)-B cell receptor and Akt Singh et al., 2007). -
Type of the Paper (Article
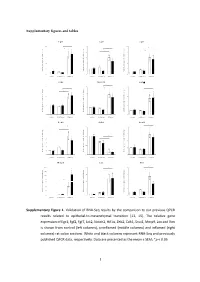
Supplementary figures and tables E g r 1 F g f2 F g f7 1 0 * 5 1 0 * * e e e * g g g * n n n * a a a 8 4 * 8 h h h * c c c d d d * l l l o o o * f f f * n n n o o o 6 3 6 i i i s s s s s s e e e r r r p p p x x x e e e 4 2 4 e e e n n n e e e g g g e e e v v v i i i t t t 2 1 2 a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d J a k 2 N o tc h 2 H if1 * 3 4 6 * * * e e e g g g n n n a a * * a * h h * h c c c 3 * d d * d l l l * o o o f f 2 f 4 n n n o o o i i i s s s s s s e e e r r 2 r p p p x x x e e e e e e n n n e e 1 e 2 g g g e e 1 e v v v i i i t t t a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Z e b 2 C d h 1 S n a i1 * * 7 1 .5 4 * * e e e g g g 6 n n n * a a a * h h h c c c 3 * d d d l l l 5 o o o f f f 1 .0 * n n n * o o o i i i 4 * s s s s s s e e e r r r 2 p p p x x x 3 e e e e e e n n n e e e 0 .5 g g g 2 e e e 1 v v v i i i t t t a a a * l l l e e e 1 * R R R 0 0 .0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d M m p 9 L o x V im 2 0 0 2 0 8 * * * e e e * g g g 1 5 0 * n n n * a a a * h h h * c c c 1 5 * 6 d d d l l l 1 0 0 o o o f f f n n n o o o i i i 5 0 s s s s s s * e e e r r r 1 0 4 3 0 p p p * x x x e e e * e e e n n n e e e 2 0 g g g e e e 5 2 v v v i i i t t t a a a l l l 1 0 e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Supplementary Figure 1. -

Ebf1-Mediated Down-Regulation of Id2 and Id3 Is Essential for Specification of the B Cell Lineage
Ebf1-mediated down-regulation of Id2 and Id3 is essential for specification of the B cell lineage Melissa A. Thala, Thiago L. Carvalhoa,TiHea, Hyung-Gyoon Kima, Hua Gaob, James Hagmanb, and Christopher A. Kluga,1 aDepartment of Microbiology, The University of Alabama-Birmingham, Birmingham, AL 35294; and bIntegrated Department of Immunology, National Jewish Medical and Research Center, Denver, CO 80206 Edited by Cornelis Murre, University of California, San Diego, La Jolla, CA, and accepted by the Editorial Board November 7, 2008 (received for review March 13, 2008) Gene knockout experiments in mice have suggested a hierarchical of E47 (13) or E12 (14) in non-B-lineage cell lines, suggest that model of early B cell commitment wherein E2A proteins (E47 and E2A activity is essential upstream of Ebf1. Similarly, the Pax5 E12) activate early B cell factor (Ebf1), which in turn activates promoter is bound by Ebf1 based on EMSA and Ebf1 can expression of the B cell commitment factor, Pax5. In IL-7 receptor transactivate the Pax5 promoter in transient co-transfection alpha (IL-7R␣) knockout mice, B cell development is blocked before assays (15, 16). Ebf1 is also present in Pax5-deficient pro-B cells B-lineage commitment at the prepro-B cell stage in adult animals. derived from Pax5Ϫ/Ϫ adult mice (17), suggesting that Ebf1 may In IL-7R␣؊/؊ prepro-B cells, E47 is expressed and yet is insufficient participate in the activation of Pax5 expression. Complicating the to transcriptionally activate the putative downstream target gene, simple hierarchical model where E2A induces expression of Ebf1. -

Functional Annotation of Human Long Noncoding Rnas Using Chromatin
bioRxiv preprint doi: https://doi.org/10.1101/2021.01.13.426305; this version posted January 14, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Funconal annotaon of human long noncoding RNAs using chroman conformaon data Saumya Agrawal1, Tanvir Alam2, Masaru Koido1,3, Ivan V. Kulakovskiy4,5, Jessica Severin1, Imad ABugessaisa1, Andrey Buyan5,6, Josee Dos&e7, Masayoshi Itoh1,8, Naoto Kondo9, Yunjing Li10, Mickaël Mendez11, Jordan A. Ramilowski1,12, Ken Yagi1, Kayoko Yasuzawa1, CHi Wai Yip1, Yasushi Okazaki1, MicHael M. Ho9man11,13,14,15, Lisa Strug10, CHung CHau Hon1, CHikashi Terao1, Takeya Kasukawa1, Vsevolod J. Makeev4,16, Jay W. SHin1, Piero Carninci1, MicHiel JL de Hoon1 1RIKEN Center for Integra&ve Medical Sciences, YokoHama, Japan. 2College of Science and Engineering, Hamad Bin KHalifa University, DoHa, Qatar. 3Ins&tute of Medical Science, THe University of Tokyo, Tokyo, Japan. 4Vavilov Ins&tute of General Gene&cs, Russian Academy of Sciences, Moscow, Russia. 5Ins&tute of Protein ResearcH, Russian Academy of Sciences, PusHcHino, Russia. 6Faculty of Bioengineering and Bioinforma&cs, Lomonosov Moscow State University, Moscow, Russia. 7Department of BiocHemistry, Rosalind and Morris Goodman Cancer ResearcH Center, McGill University, Montréal, QuéBec, Canada. 8RIKEN Preven&ve Medicine and Diagnosis Innova&on Program, Wako, Japan. 9RIKEN Center for Life Science TecHnologies, YokoHama, Japan. 10Division of Biosta&s&cs, Dalla Lana ScHool of PuBlic HealtH, University of Toronto, Toronto, Ontario, Canada. 11Department of Computer Science, University of Toronto, Toronto, Ontario, Canada. 12Advanced Medical ResearcH Center, YokoHama City University, YokoHama, Japan.