HSF1) During Macrophage Differentiation of Monocytes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Role of PU.1 and GATA-1 Transcription Factors During Normal and Leukemogenic Hematopoiesis
Leukemia (2010) 24, 1249–1257 & 2010 Macmillan Publishers Limited All rights reserved 0887-6924/10 www.nature.com/leu REVIEW The role of PU.1 and GATA-1 transcription factors during normal and leukemogenic hematopoiesis P Burda1, P Laslo2 and T Stopka1,3 1Department of Pathophysiology and Center of Experimental Hematology, First Faculty of Medicine, Charles University, Prague, Czech Republic; 2Section of Experimental Haematology, Leeds Institute of Molecular Medicine, University of Leads, St James’s University Hospital, Leeds, UK and 31st Department of Medicine-Hematology, General University Hospital, Prague, Czech Republic Hematopoiesis is coordinated by a complex regulatory network Additional domains include an N-terminal acidic domain and a of transcription factors and among them PU.1 (Spi1, Sfpi1) glutamine-rich domain, both involved in transcriptional activa- represents a key molecule. This review summarizes the tion, as well as a PEST domain involved in protein–protein indispensable requirement of PU.1 during hematopoietic cell fate decisions and how the function of PU.1 can be modulated interactions. PU.1 protein can be modified post-translationally by protein–protein interactions with additional factors. The by phosporylation at serines 41 (N-terminal acidic domain) and mutual negative regulation between PU.1 and GATA-1 is 142 and 148 (PEST domain), which results in augmented detailed within the context of normal and leukemogenic activity. hematopoiesis and the concept of ‘differentiation therapy’ to The PU.1 protein can physically interact with a variety of restore normal cellular differentiation of leukemic cells is regulatory factors including (i) general transcription factors discussed. Leukemia (2010) 24, 1249–1257; doi:10.1038/leu.2010.104; (TFIID, TBP), (ii) early hematopoietic transcription factors published online 3 June 2010 (GATA-2 and Runx-1), (iii) erythroid factor (GATA-1) and (iv) Keywords: PU.1; leukemia differentiation; GATA-1; chromatin; non-erythroid factors (C/EBPa, C/EBPb, IRF4/8 and c-Jun). -

Activated Peripheral-Blood-Derived Mononuclear Cells
Transcription factor expression in lipopolysaccharide- activated peripheral-blood-derived mononuclear cells Jared C. Roach*†, Kelly D. Smith*‡, Katie L. Strobe*, Stephanie M. Nissen*, Christian D. Haudenschild§, Daixing Zhou§, Thomas J. Vasicek¶, G. A. Heldʈ, Gustavo A. Stolovitzkyʈ, Leroy E. Hood*†, and Alan Aderem* *Institute for Systems Biology, 1441 North 34th Street, Seattle, WA 98103; ‡Department of Pathology, University of Washington, Seattle, WA 98195; §Illumina, 25861 Industrial Boulevard, Hayward, CA 94545; ¶Medtronic, 710 Medtronic Parkway, Minneapolis, MN 55432; and ʈIBM Computational Biology Center, P.O. Box 218, Yorktown Heights, NY 10598 Contributed by Leroy E. Hood, August 21, 2007 (sent for review January 7, 2007) Transcription factors play a key role in integrating and modulating system. In this model system, we activated peripheral-blood-derived biological information. In this study, we comprehensively measured mononuclear cells, which can be loosely termed ‘‘macrophages,’’ the changing abundances of mRNAs over a time course of activation with lipopolysaccharide (LPS). We focused on the precise mea- of human peripheral-blood-derived mononuclear cells (‘‘macro- surement of mRNA concentrations. There is currently no high- phages’’) with lipopolysaccharide. Global and dynamic analysis of throughput technology that can precisely and sensitively measure all transcription factors in response to a physiological stimulus has yet to mRNAs in a system, although such technologies are likely to be be achieved in a human system, and our efforts significantly available in the near future. To demonstrate the potential utility of advanced this goal. We used multiple global high-throughput tech- such technologies, and to motivate their development and encour- nologies for measuring mRNA levels, including massively parallel age their use, we produced data from a combination of two distinct signature sequencing and GeneChip microarrays. -

The Unique Transcriptional Response Produced by Concurrent Estrogen
Need et al. BMC Cancer (2015) 15:791 DOI 10.1186/s12885-015-1819-3 RESEARCH ARTICLE Open Access The unique transcriptional response produced by concurrent estrogen and progesterone treatment in breast cancer cells results in upregulation of growth factor pathways and switching from a Luminal A to a Basal-like subtype Eleanor F. Need1*,LukeA.Selth2,3,AndrewP.Trotta1,4,DamienA.Leach1,LaurenGiorgio1, Melissa A. O’Loughlin1, Eric Smith5, Peter G. Gill6,WendyV.Ingman7,8, J. Dinny Graham9 and Grant Buchanan1,3 Abstract Background: In breast cancer, progesterone receptor (PR) positivity or abundance is positively associated with survival and treatment response. It was initially believed that PR was a useful diagnostic marker of estrogen receptor activity, but increasingly PR has been recognised to play an important biological role in breast homeostasis, carcinogenesis and metastasis. Although PR expression is almost exclusively observed in estrogen receptor positive tumors, few studies have investigated the cellular mechanisms of PR action in the context of ongoing estrogen signalling. Methods: In this study, we contrast PR function in estrogen pretreated ZR-75-1 breast cancer cells with vehicle treated ZR-75-1 and T-47D breast cancer cells using expression microarrays and chromatin immunoprecipitation-sequencing. Results: Estrogen cotreatment caused a dramatic increase in the number of genes regulated by progesterone in ZR-75-1 cells. In T-47D cells that have naturally high levels of PR, estrogen and progesterone cotreatment resulted in a reduction in the number of regulated genes in comparison to treatment with either hormone alone. At a genome level, estrogen pretreatment of ZR-75-1 cells led to a 10-fold increase in the number of PR DNA binding sites detected using ChIP-sequencing. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Heat Shock Factor 1 Mediates Latent HIV Reactivation
www.nature.com/scientificreports OPEN Heat Shock Factor 1 Mediates Latent HIV Reactivation Xiao-Yan Pan1,*, Wei Zhao1,2,*, Xiao-Yun Zeng1, Jian Lin1, Min-Min Li3, Xin-Tian Shen1 & Shu-Wen Liu1,2 Received: 19 October 2015 HSF1, a conserved heat shock factor, has emerged as a key regulator of mammalian transcription Accepted: 29 April 2016 in response to cellular metabolic status and stress. To our knowledge, it is not known whether Published: 18 May 2016 HSF1 regulates viral transcription, particularly HIV-1 and its latent form. Here we reveal that HSF1 extensively participates in HIV transcription and is critical for HIV latent reactivation. Mode of action studies demonstrated that HSF1 binds to the HIV 5′-LTR to reactivate viral transcription and recruits a family of closely related multi-subunit complexes, including p300 and p-TEFb. And HSF1 recruits p300 for self-acetylation is also a committed step. The knockout of HSF1 impaired HIV transcription, whereas the conditional over-expression of HSF1 improved that. These findings demonstrate that HSF1 positively regulates the transcription of latent HIV, suggesting that it might be an important target for different therapeutic strategies aimed at a cure for HIV/AIDS. The long-lived latent viral reservoir of HIV-1 prevents its eradication and the development of a cure1. The recent combination antiretroviral therapy (cART) aimed at inhibiting viral enzymatic activities prevents HIV-1 repli- cation and halts the viral destruction of the host immune system2. However, proviruses in the latent reservoir persist in a transcriptionally inactive state, are insuppressible by cART and undetectable by the immune system3. -

HSF1-Mediated Control of Cellular Energy Metabolism and Mtorc1 Activation
Author Manuscript Published OnlineFirst on November 19, 2019; DOI: 10.1158/1541-7786.MCR-19-0217 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. HSF1-mediated Control of Cellular Energy Metabolism and mTORC1 Activation Drive Acute T Cell Lymphoblastic Leukemia Progression Binnur Eroglu1, Junfeng Pang1, Xiongjie Jin1, Caixia Xi1, Demetrius Moskophidis1,2 Nahid F. Mivechi1,2, 3 1Molecular Chaperone Biology, Medical College of Georgia, Georgia Cancer Center, 2Department of Medicine, and 3Department of Radiation Oncology, Augusta University, Augusta, Georgia, USA. B. Eroglu and J. Pang contributed equally to this article. Running Title: Increased susceptibility of T-ALL to HSF1 depletion Corresponding Authors: Nahid F. Mivechi, Augusta University, Augusta, 1120 15th St., CN-3153, Augusta, Georgia, 30912; Phone: 706-721-8759; E-mail: [email protected]; and Demetrius Moskophidis, Augusta University, Augusta, 1120 15th St., CN-3143, Augusta, Georgia, 30912; Phone: 706-721-8738; E- mail: [email protected]. Disclosure of potential conflicts of interest: Authors declare no potential conflict of interest. 1 Downloaded from mcr.aacrjournals.org on October 3, 2021. © 2019 American Association for Cancer Research. Author Manuscript Published OnlineFirst on November 19, 2019; DOI: 10.1158/1541-7786.MCR-19-0217 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Abstract Deregulated oncogenic signaling linked to PI3K/AKT and mTORC1 pathway activation is a hallmark of human T cell acute leukemia (T-ALL) pathogenesis and contributes to leukemic cell resistance and adverse prognosis. Notably, although the multi-agent chemotherapy of leukemia leads to a high rate of complete remission, options for salvage therapy for relapsed/refractory disease are limited due to the serious side effects of augmenting cytotoxic chemotherapy. -

Development and Validation of a Novel Immune-Related Prognostic Model
Wang et al. J Transl Med (2020) 18:67 https://doi.org/10.1186/s12967-020-02255-6 Journal of Translational Medicine RESEARCH Open Access Development and validation of a novel immune-related prognostic model in hepatocellular carcinoma Zheng Wang1, Jie Zhu1, Yongjuan Liu3, Changhong Liu2, Wenqi Wang2, Fengzhe Chen1* and Lixian Ma1* Abstract Background: Growing evidence has suggested that immune-related genes play crucial roles in the development and progression of hepatocellular carcinoma (HCC). Nevertheless, the utility of immune-related genes for evaluating the prognosis of HCC patients are still lacking. The study aimed to explore gene signatures and prognostic values of immune-related genes in HCC. Methods: We comprehensively integrated gene expression data acquired from 374 HCC and 50 normal tissues in The Cancer Genome Atlas (TCGA). Diferentially expressed genes (DEGs) analysis and univariate Cox regression analysis were performed to identify DEGs that related to overall survival. An immune prognostic model was constructed using the Lasso and multivariate Cox regression analyses. Furthermore, Cox regression analysis was applied to identify independent prognostic factors in HCC. The correlation analysis between immune-related signature and immune cells infltration were also investigated. Finally, the signature was validated in an external independent dataset. Results: A total of 329 diferentially expressed immune‐related genes were detected. 64 immune‐related genes were identifed to be markedly related to overall survival in HCC patients using univariate Cox regression analysis. Then we established a TF-mediated network for exploring the regulatory mechanisms of these genes. Lasso and multivariate Cox regression analyses were applied to construct the immune-based prognostic model, which consisted of nine immune‐related genes. -

HSF1 in the Yeast Saccharomyces Cerevisiaeã
Mol Gen Genet (1997) 255:322±331 Ó Springer-Verlag 1997 ORIGINAL PAPER O. Boscheinen á R. Lyck á C. Queitsch á E. Treuter V. Zimarino á K.-D. Scharf Heat stress transcription factors from tomato can functionally replace HSF1 in the yeast Saccharomyces cerevisiaeà Received: 1 October 1996 / Accepted: 6 December 1996 Abstract The fact that yeast HSF1 is essential for sur- nomeric Hsf has a markedly reduced anity for DNA, as vival under nonstress conditions can be used to test shown by lacZ reporter and band-shift assays. heterologous Hsfs for the ability to substitute for the endogenous protein. Our results demonstrate that like Key words Tomato á Yeast á Heat stress á Transcription Hsf of Drosophila, tomato Hsfs A1 and A2 can func- factor á Thermotolerance tionally replace the corresponding yeast protein, but Hsf B1 cannot. In addition to survival at 28° C, we checked the transformed yeast strains for temperature sensitivity Introduction of growth, induced thermotolerance and activator func- tion using two dierent lacZ reporter constructs. Tests The striking conservation of essential elements of the with full-length Hsfs were supplemented by assays using heat stress response is well documented by the 11 heat mutant Hsfs lacking parts of their C-terminal activator stress protein (Hsp) families, with representatives found region or oligomerization domain, or containing amino in all organisms investigated so far. As molecular acid substitutions in the DNA-binding domain. Re- chaperones, they form part of a homeostatic network markably, results with the yeast system are basically responsible for the proper folding, assembly, intracellu- similar to those obtained by the analysis of the same Hsfs lar distribution and degradation of proteins (see sum- as transcriptional activators in a tobacco protoplast as- maries by Nover 1991; Buchner 1996; Hartl 1996; say. -

Single-Cell RNA Sequencing Analysis of Human Neural Grafts Revealed Unexpected Cell Type Underlying the Genetic Risk of Parkinson’S Disease
Single-cell RNA Sequencing Analysis of Human Neural Grafts Revealed Unexpected Cell Type Underlying the Genetic Risk of Parkinson’s Disease Yingshan Wang1, Gang Wu2 1 Episcopal High School, 1200 N Quaker Ln, Alexandria, VA, USA, 22302 2 Fujian Sanbo Funeng Brain Hospital; Sanbo Brain Hospital Capital Medical University Abstract Parkinson’s disease (PD) is the second most common neurodegenerative disorder, affecting more than 6 million patients globally. Though previous studies have proposed several disease-related molecular pathways, how cell-type specific mechanisms contribute to the pathogenesis of PD is still mostly unknown. In this study, we analyzed single-cell RNA sequencing data of human neural grafts transplanted to the midbrains of rat PD models. Specifically, we performed cell-type identification, risk gene screening, and co-expression analysis. Our results revealed the unexpected genetic risk of oligodendrocytes as well as important pathways and transcription factors in PD pathology. The study may provide an overarching framework for understanding the cell non- autonomous effects in PD, inspiring new research hypotheses and therapeutic strategies. Keywords Parkinson’s Disease; Single-cell RNA Sequencing; Oligodendrocytes; Cell Non-autonomous; Co- expression Analysis; Transcription Factors 1 Table of Contents 1. Introduction ................................................................................................................................. 3 2. Methods...................................................................................................................................... -
Type of the Paper (Article
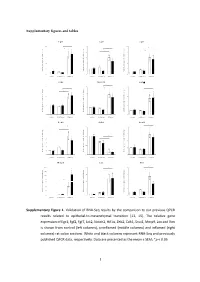
Supplementary figures and tables E g r 1 F g f2 F g f7 1 0 * 5 1 0 * * e e e * g g g * n n n * a a a 8 4 * 8 h h h * c c c d d d * l l l o o o * f f f * n n n o o o 6 3 6 i i i s s s s s s e e e r r r p p p x x x e e e 4 2 4 e e e n n n e e e g g g e e e v v v i i i t t t 2 1 2 a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d J a k 2 N o tc h 2 H if1 * 3 4 6 * * * e e e g g g n n n a a * * a * h h * h c c c 3 * d d * d l l l * o o o f f 2 f 4 n n n o o o i i i s s s s s s e e e r r 2 r p p p x x x e e e e e e n n n e e 1 e 2 g g g e e 1 e v v v i i i t t t a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Z e b 2 C d h 1 S n a i1 * * 7 1 .5 4 * * e e e g g g 6 n n n * a a a * h h h c c c 3 * d d d l l l 5 o o o f f f 1 .0 * n n n * o o o i i i 4 * s s s s s s e e e r r r 2 p p p x x x 3 e e e e e e n n n e e e 0 .5 g g g 2 e e e 1 v v v i i i t t t a a a * l l l e e e 1 * R R R 0 0 .0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d M m p 9 L o x V im 2 0 0 2 0 8 * * * e e e * g g g 1 5 0 * n n n * a a a * h h h * c c c 1 5 * 6 d d d l l l 1 0 0 o o o f f f n n n o o o i i i 5 0 s s s s s s * e e e r r r 1 0 4 3 0 p p p * x x x e e e * e e e n n n e e e 2 0 g g g e e e 5 2 v v v i i i t t t a a a l l l 1 0 e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Supplementary Figure 1. -

Supplementary Table 5. Clover Results Indicate the Number Of
Supplementary Table 5. Clover results indicate the number of chromosomes with transcription factor binding motifs statistically over‐ or under‐represented in HTE DHS within intergenic sequence (more than 2kb outside of any gene). Analysis was divided into three groups (all DHS, HTE‐selective DHS, and ubiquitous DHS). Motifs with more than one entry in the databases utilized were edited to retain only the first occurrence of the motif. All DHS x Intergenic TEselective DHS x Intergenic Ubiquitous DHS x Intergenic ID Name p < 0.01 p > 0.99 ID Name p < 0.01 p > 0.99 ID Name p < 0.01 p > 0.99 MA0002.2 RUNX1 23 0 MA0080.2 SPI1 23 0 MA0055.1 Myf 23 0 MA0003.1 TFAP2A 23 0 MA0089.1 NFE2L1::MafG 23 0 MA0068.1 Pax4 23 0 MA0039.2 Klf4 23 0 MA0098.1 ETS1 23 0 MA0080.2 SPI1 23 0 MA0055.1 Myf 23 0 MA0099.2 AP1 23 0 MA0098.1 ETS1 23 0 MA0056.1 MZF1_1‐4 23 0 MA0136.1 ELF5 23 0 MA0139.1 CTCF 23 0 MA0079.2 SP1 23 0 MA0145.1 Tcfcp2l1 23 0 V$ALX3_01 ALX‐3 23 0 MA0080.2 SPI1 23 0 MA0150.1 NFE2L2 23 0 V$ALX4_02 Alx‐4 23 0 myocyte enhancer MA0081.1 SPIB 23 0 MA0156.1 FEV 23 0 V$AMEF2_Q6 factor 23 0 MA0089.1 NFE2L1::MafG 23 0 V$AP1FJ_Q2 activator protein 1 23 0 V$AP1_01 AP‐1 23 0 MA0090.1 TEAD1 23 0 V$AP4_Q5 activator protein 4 23 0 V$AP2_Q6_01 AP‐2 23 0 MA0098.1 ETS1 23 0 V$AR_Q6 half‐site matrix 23 0 V$ARX_01 Arx 23 0 BTB and CNC homolog MA0099.2 AP1 23 0 V$BACH1_01 1 23 0 V$BARHL1_01 Barhl‐1 23 0 BTB and CNC homolog MA0136.1 ELF5 23 0 V$BACH2_01 2 23 0 V$BARHL2_01 Barhl2 23 0 MA0139.1 CTCF 23 0 V$CMAF_02 C‐MAF 23 0 V$BARX1_01 Barx1 23 0 MA0144.1 Stat3 23 0 -

What Your Genome Doesn't Tell
UC San Diego UC San Diego Electronic Theses and Dissertations Title Multi-layered epigenetic control of T cell fate decisions Permalink https://escholarship.org/uc/item/8rs7c7b3 Author Yu, Bingfei Publication Date 2018 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA SAN DIEGO Multi-layered epigenetic control of T cell fate decisions A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Biology by Bingfei Yu Committee in charge: Professor Ananda Goldrath, Chair Professor John Chang Professor Stephen Hedrick Professor Cornelis Murre Professor Wei Wang 2018 Copyright Bingfei Yu, 2018 All rights reserved. The dissertation of Bingfei Yu is approved, and it is ac- ceptable in quality and form for publication on microfilm and electronically: Chair University of California San Diego 2018 iii DEDICATION To my parents who have been giving me countless love, trust and support to make me who I am. iv EPIGRAPH Stay hungary. Stay foolish. | Steve Jobs quoted from the back cover of the 1974 edition of the Whole Earth Catalog v TABLE OF CONTENTS Signature Page.................................. iii Dedication..................................... iv Epigraph.....................................v Table of Contents................................. vi List of Figures.................................. ix Acknowledgements................................x Vita........................................ xii Abstract of