Cardiac Fibroblast P38 MAPK: a Critical Regulator of Myocardial Remodeling
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

N-Glycan Trimming in the ER and Calnexin/Calreticulin Cycle
Neurotransmitter receptorsGABA and A postsynapticreceptor activation signal transmission Ligand-gated ion channel transport GABAGABA Areceptor receptor alpha-5 alpha-1/beta-1/gamma-2 subunit GABA A receptor alpha-2/beta-2/gamma-2GABA receptor alpha-4 subunit GABAGABA receptor A receptor beta-3 subunitalpha-6/beta-2/gamma-2 GABA-AGABA receptor; A receptor alpha-1/beta-2/gamma-2GABA receptoralpha-3/beta-2/gamma-2 alpha-3 subunit GABA-A GABAreceptor; receptor benzodiazepine alpha-6 subunit site GABA-AGABA-A receptor; receptor; GABA-A anion site channel (alpha1/beta2 interface) GABA-A receptor;GABA alpha-6/beta-3/gamma-2 receptor beta-2 subunit GABAGABA receptorGABA-A receptor alpha-2receptor; alpha-1 subunit agonist subunit GABA site Serotonin 3a (5-HT3a) receptor GABA receptorGABA-C rho-1 subunitreceptor GlycineSerotonin receptor subunit3 (5-HT3) alpha-1 receptor GABA receptor rho-2 subunit GlycineGlycine receptor receptor subunit subunit alpha-2 alpha-3 Ca2+ activated K+ channels Metabolism of ingested SeMet, Sec, MeSec into H2Se SmallIntermediateSmall conductance conductance conductance calcium-activated calcium-activated calcium-activated potassium potassium potassiumchannel channel protein channel protein 2 protein 1 4 Small conductance calcium-activatedCalcium-activated potassium potassium channel alpha/beta channel 1 protein 3 Calcium-activated potassiumHistamine channel subunit alpha-1 N-methyltransferase Neuraminidase Pyrimidine biosynthesis Nicotinamide N-methyltransferase Adenosylhomocysteinase PolymerasePolymeraseHistidine basic -

YAP and TAZ Mediators at the Crossroad Between Metabolic and Cellular Reprogramming
H OH metabolites OH Review YAP and TAZ Mediators at the Crossroad between Metabolic and Cellular Reprogramming Giorgia Di Benedetto, Silvia Parisi , Tommaso Russo and Fabiana Passaro * Department of Molecular Medicine and Medical Biotechnology, University of Naples Federico II, 40, 80138 Napoli, Italy; [email protected] (G.D.B.); [email protected] (S.P.); [email protected] (T.R.) * Correspondence: [email protected] Abstract: Cell reprogramming can either refer to a direct conversion of a specialized cell into another or to a reversal of a somatic cell into an induced pluripotent stem cell (iPSC). It implies a peculiar modification of the epigenetic asset and gene regulatory networks needed for a new cell, to better fit the new phenotype of the incoming cell type. Cellular reprogramming also implies a metabolic rearrangement, similar to that observed upon tumorigenesis, with a transition from oxidative phosphorylation to aerobic glycolysis. The induction of a reprogramming process requires a nexus of signaling pathways, mixing a range of local and systemic information, and accumulating evidence points to the crucial role exerted by the Hippo pathway components Yes-Associated Protein (YAP) and Transcriptional Co-activator with PDZ-binding Motif (TAZ). In this review, we will first provide a synopsis of the Hippo pathway and its function during reprogramming and tissue regeneration, then we introduce the latest knowledge on the interplay between YAP/TAZ Citation: Di Benedetto, G.; Parisi, S.; and metabolism and, finally, we discuss the possible role of YAP/TAZ in the orchestration of the Russo, T.; Passaro, F. YAP and TAZ metabolic switch upon cellular reprogramming. -

IGF2BP1 Is a Targetable SRC/MAPK-Dependent Driver Of
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.19.159905; this version posted June 20, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 IGF2BP1 is a targetable SRC/MAPK-dependent driver of invasive growth in ovarian 2 cancer 3 4 Authors: Nadine Bley1*$#, Annekatrin Schott1*, Simon Müller1, Danny Misiak1, Marcell 5 Lederer1, Tommy Fuchs1, Chris Aßmann1, Markus Glaß1, Christian Ihling2, Andrea Sinz2, 6 Nikolaos Pazaitis3, Claudia Wickenhauser3, Martina Vetter4, Olga Ungurs4, Hans-Georg 7 Strauss4, Christoph Thomssen4, Stefan Hüttelmaier1$. 8 9 10 Addresses: 11 1 Sect. Molecular Cell Biology, Inst. of Molecular Medicine, Charles Tanford Protein Center, 12 Medical Faculty, Martin Luther University Halle-Wittenberg, Kurt-Mothes-Str. 3A, 06120 Halle, 13 Germany 14 2 Dept. of Pharmaceutical Chemistry & Bioanalytics, Inst. of Pharmacy, Charles Tanford 15 Protein Center, Martin Luther University Halle-Wittenberg, Kurt-Mothes Str. 3A, 06120 Halle, 16 Germany 17 3 Inst. of Pathology, Medical Faculty, Martin Luther University Halle-Wittenberg, Magdeburger 18 Str. 14, 06112 Halle, Germany 19 4 Clinics for Gynecology, Medical Faculty, Martin Luther University Halle-Wittenberg, Ernst- 20 Grube-Str. 40, 06120 Halle, Germany 21 22 23 * These authors contributed equally to this work 24 $ shared corresponding authorship 25 # Correspondence: should be addressed to Nadine Bley 26 Running title: IGF2BP1-directed AJ disassembly depends on SRC activation 27 Disclosure of Potential Conflicts of Interests: the authors declare no conflicts of interest. -

MAPK4 Overexpression Promotes Tumor Progression Via Noncanonical Activation of AKT/Mtor Signaling
The Journal of Clinical Investigation RESEARCH ARTICLE MAPK4 overexpression promotes tumor progression via noncanonical activation of AKT/mTOR signaling Wei Wang,1 Tao Shen,1 Bingning Dong,1 Chad J. Creighton,2,3 Yanling Meng,1 Wolong Zhou,1 Qing Shi,1 Hao Zhou,1 Yinjie Zhang,1 David D. Moore,1 and Feng Yang1 1Department of Molecular and Cellular Biology, 2Department of Medicine, and 3Dan L. Duncan Cancer Center, Baylor College of Medicine, Houston, Texas, USA. MAPK4 is an atypical MAPK. Currently, little is known about its physiological function and involvement in diseases, including cancer. A comprehensive analysis of 8887 gene expression profiles in The Cancer Genome Atlas (TCGA) revealed that MAPK4 overexpression correlates with decreased overall survival, with particularly marked survival effects in patients with lung adenocarcinoma, bladder cancer, low-grade glioma, and thyroid carcinoma. Interestingly, human tumor MAPK4 overexpression also correlated with phosphorylation of AKT, 4E-BP1, and p70S6K, independent of the loss of PTEN or mutation of PIK3CA. This led us to examine whether MAPK4 activates the key metabolic, prosurvival, and proliferative kinase AKT and mTORC1 signaling, independent of the canonical PI3K pathway. We found that MAPK4 activated AKT via a novel, concerted mechanism independent of PI3K. Mechanistically, MAPK4 directly bound and activated AKT by phosphorylation of the activation loop at threonine 308. It also activated mTORC2 to phosphorylate AKT at serine 473 for full activation. MAPK4 overexpression induced oncogenic outcomes, including transforming prostate epithelial cells into anchorage-independent growth, and MAPK4 knockdown inhibited cancer cell proliferation, anchorage-independent growth, and xenograft growth. We concluded that MAPK4 can promote cancer by activating the AKT/mTOR signaling pathway and that targeting MAPK4 may provide a novel therapeutic approach for cancer. -

HHS Public Access Author Manuscript
HHS Public Access Author manuscript Author Manuscript Author ManuscriptBreast Cancer Author Manuscript Res Treat Author Manuscript . Author manuscript; available in PMC 2016 June 01. Published in final edited form as: Breast Cancer Res Treat. 2015 June ; 151(2): 453–463. doi:10.1007/s10549-015-3401-8. Body mass index associated with genome-wide methylation in breast tissue Brionna Y. Hair1, Zongli Xu2, Erin L. Kirk1, Sophia Harlid2, Rupninder Sandhu3, Whitney R. Robinson1,3, Michael C. Wu4, Andrew F. Olshan1, Kathleen Conway1,3, Jack A. Taylor2, and Melissa A. Troester1 1 Department of Epidemiology, University of North Carolina at Chapel Hill, CB #7435, 2101 McGavran-Greenberg Hall, Chapel Hill, NC 27599-7435, USA 2 Epidemiology Branch, and Epigenomics and Stem Cell Biology Laboratory, National Institute of Environmental Health Sciences (NIH), Research Triangle Park, NC, USA 3 Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA 4 Fred Hutchinson Cancer Research Center, Seattle, WA, USA Abstract Gene expression studies indicate that body mass index (BMI) is associated with molecular pathways involved in inflammation, insulin-like growth factor activation, and other carcinogenic processes in breast tissue. The goal of this study was to determine whether BMI is associated with gene methylation in breast tissue and to identify pathways that are commonly methylated in association with high BMI. Epigenome-wide methylation profiles were determined using the Illumina HumanMethylation450 BeadChip array in the non-diseased breast tissue of 81 women undergoing breast surgery between 2009 and 2013 at the University of North Carolina Hospitals. Multivariable, robust linear regression was performed to identify methylation sites associated with BMI at a false discovery rate q value <0.05. -
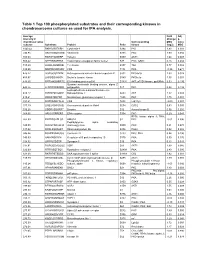
Table 1 Top 100 Phosphorylated Substrates and Their Corresponding Kinases in Chondrosarcoma Cultures As Used for IPA Analysis
Table 1 Top 100 phosphorylated substrates and their corresponding kinases in chondrosarcoma cultures as used for IPA analysis. Average Fold Adj intensity in Change p- chondrosarcoma Corresponding MSC value cultures Substrate Protein Psite kinase (log2) MSC 1043.42 RKKKVSSTKRH Cytohesin-1 S394 PKC 1.83 0.001 746.95 RKGYRSQRGHS Vitronectin S381 PKC 1.00 0.056 709.03 RARSTSLNERP Tuberin S939 AKT1 1.64 0.008 559.42 SPPRSSLRRSS Transcription elongation factor A-like1 S37 PKC; GSK3 0.18 0.684 515.29 LRRSLSRSMSQ Telethonin S157 Titin 0.77 0.082 510.00 MQPDNSSDSDY CD5 T434 PKA -0.35 0.671 476.27 GGRGGSRARNL Heterogeneous nuclear ribonucleoprotein K S302 PKCdelta 1.03 0.028 455.97 LKPGSSHRKTK Bruton's tyrosine kinase S180 PKCbeta 1.55 0.001 444.65 RRRMASMQRTG E1A binding protein p300 S1834 AKT; p70S6 kinase; pp90Rsk 0.53 0.195 Guanine nucleotide binding protein, alpha Z 440.26 HLRSESQRQRR polypeptide S27 PKC 0.88 0.199 6-phosphofructo-2-kinase/fructose-2,6- 424.12 RPRNYSVGSRP biphosphatase 2 S483 AKT 1.32 0.003 419.61 KKKIATRKPRF Metabotropic glutamate receptor 1 T695 PKC 1.75 0.001 391.21 DNSSDSDYDLH CD5 T453 Lck; Fyn -2.09 0.001 377.39 LRQLRSPRRAQ Ras associated protein Rab4 S204 CDC2 0.63 0.091 376.28 SSQRVSSYRRT Desmin S12 Aurora kinase B 0.56 0.255 369.05 ARIGGSRRERS EP4 receptor S354 PKC 0.29 0.543 RPS6 kinase alpha 3; PKA; 367.99 EPKRRSARLSA HMG14 S7 PKC -0.01 0.996 Peptidylglycine alpha amidating 349.08 SRKGYSRKGFD monooxygenase S930 PKC 0.21 0.678 347.92 RRRLSSLRAST Ribosomal protein S6 S236 PAK2 0.02 0.985 346.84 RSNPPSRKGSG Connexin -

Apoptosis Signal-Regulating Kinase 1 Promotes Ochratoxin A-Induced
OPEN Apoptosis Signal-regulating Kinase 1 SUBJECT AREAS: promotes Ochratoxin A-induced renal CELL BIOLOGY RISK FACTORS cytotoxicity Rui Liang1, Xiao Li Shen1,2, Boyang Zhang1, Yuzhe Li1, Wentao Xu1, Changhui Zhao3, YunBo Luo1 Received & Kunlun Huang1 10 November 2014 Accepted 1Laboratory of food safety and molecular biology, College of Food Science and Nutritional Engineering, China Agricultural 5 January 2015 University, Beijing 100083, P.R. China, 2School of Public Health, Zunyi Medical University, Zunyi, Guizhou 563003, P.R. China, 3Department of Nutrition and Food Science, University of Maryland, College Park, MD 20742, USA. Published 28 January 2015 Oxidative stress and apoptosis are involved in Ochratoxin A (OTA)-induced renal cytotoxicity. Apoptosis signal-regulating kinase 1 (ASK1) is a Mitogen-Activated Protein Kinase Kinase Kinase (MAPKKK, Correspondence and MAP3K) family member that plays an important role in oxidative stress-induced cell apoptosis. In this study, we performed RNA interference of ASK1 in HEK293 cells and employed an iTRAQ-based requests for materials quantitative proteomics approach to globally investigate the regulatory mechanism of ASK1 in should be addressed to OTA-induced renal cytotoxicity. Our results showed that ASK1 knockdown alleviated OTA-induced ROS W.X. (xuwentao@cau. generation and Dym loss and thus desensitized the cells to OTA-induced apoptosis. We identified 33 and 24 edu.cn) differentially expressed proteins upon OTA treatment in scrambled and ASK1 knockdown cells, respectively. Pathway classification and analysis revealed that ASK1 participated in OTA-induced inhibition of mRNA splicing, nucleotide metabolism, the cell cycle, DNA repair, and the activation of lipid metabolism. We concluded that ASK1 plays an essential role in promoting OTA-induced renal cytotoxicity. -

Overexpression of an Activated REL Mutant Enhances the Transformed State of the Human B-Lymphoma BJAB Cell Line and Alters Its Gene Expression Profile
Oncogene (2009) 28, 2100–2111 & 2009 Macmillan Publishers Limited All rights reserved 0950-9232/09 $32.00 www.nature.com/onc ORIGINAL ARTICLE Overexpression of an activated REL mutant enhances the transformed state of the human B-lymphoma BJAB cell line and alters its gene expression profile M Chin1, M Herscovitch1, N Zhang, DJ Waxman and TD Gilmore Department of Biology, Boston University, Boston, MA, USA The human REL proto-oncogene encodes a transcription factor. Misregulated REL is associated with B-cell factor in the nuclear factor (NF)-kB family. Overexpres- malignancies in several ways (Gilmore et al., sion of REL is acutely transforming in chicken lymphoid 2004). Overexpression of REL protein can transform cells, but has not been shown to transform any mammalian chicken lymphoid cells in vitro. Additionally, the lymphoid cell type. In this report, we show that over- REL locus is amplified in several types of human expression of a highly transforming mutant of REL B-cell lymphoma, including diffuse large B-cell lympho- (RELDTAD1) increases the oncogenic properties of the ma (DLBCL), follicular and primary mediastinal human B-cell lymphoma BJAB cell line, as shown by lymphomas. Moreover, REL mRNA is highly expressed increased colony formation in soft agar, tumor formation in de novo DLBCLs, and this elevated expression in SCID (severe combined immunodeficient) mice, and correlates with increased expression of many adhesion. BJAB-RELDTAD1 cells also show decreased putative REL target genes (Rhodes et al., 2005). activation of caspase in response to doxorubicin. BJAB- Nevertheless, REL has not been shown to be oncogenic RELDTAD1 cells have increased levels of active nuclear in any mammalian B-cell system, either in vitro REL protein as determined by immunofluorescence, or in vivo. -

Xo PANEL DNA GENE LIST
xO PANEL DNA GENE LIST ~1700 gene comprehensive cancer panel enriched for clinically actionable genes with additional biologically relevant genes (at 400 -500x average coverage on tumor) Genes A-C Genes D-F Genes G-I Genes J-L AATK ATAD2B BTG1 CDH7 CREM DACH1 EPHA1 FES G6PC3 HGF IL18RAP JADE1 LMO1 ABCA1 ATF1 BTG2 CDK1 CRHR1 DACH2 EPHA2 FEV G6PD HIF1A IL1R1 JAK1 LMO2 ABCB1 ATM BTG3 CDK10 CRK DAXX EPHA3 FGF1 GAB1 HIF1AN IL1R2 JAK2 LMO7 ABCB11 ATR BTK CDK11A CRKL DBH EPHA4 FGF10 GAB2 HIST1H1E IL1RAP JAK3 LMTK2 ABCB4 ATRX BTRC CDK11B CRLF2 DCC EPHA5 FGF11 GABPA HIST1H3B IL20RA JARID2 LMTK3 ABCC1 AURKA BUB1 CDK12 CRTC1 DCUN1D1 EPHA6 FGF12 GALNT12 HIST1H4E IL20RB JAZF1 LPHN2 ABCC2 AURKB BUB1B CDK13 CRTC2 DCUN1D2 EPHA7 FGF13 GATA1 HLA-A IL21R JMJD1C LPHN3 ABCG1 AURKC BUB3 CDK14 CRTC3 DDB2 EPHA8 FGF14 GATA2 HLA-B IL22RA1 JMJD4 LPP ABCG2 AXIN1 C11orf30 CDK15 CSF1 DDIT3 EPHB1 FGF16 GATA3 HLF IL22RA2 JMJD6 LRP1B ABI1 AXIN2 CACNA1C CDK16 CSF1R DDR1 EPHB2 FGF17 GATA5 HLTF IL23R JMJD7 LRP5 ABL1 AXL CACNA1S CDK17 CSF2RA DDR2 EPHB3 FGF18 GATA6 HMGA1 IL2RA JMJD8 LRP6 ABL2 B2M CACNB2 CDK18 CSF2RB DDX3X EPHB4 FGF19 GDNF HMGA2 IL2RB JUN LRRK2 ACE BABAM1 CADM2 CDK19 CSF3R DDX5 EPHB6 FGF2 GFI1 HMGCR IL2RG JUNB LSM1 ACSL6 BACH1 CALR CDK2 CSK DDX6 EPOR FGF20 GFI1B HNF1A IL3 JUND LTK ACTA2 BACH2 CAMTA1 CDK20 CSNK1D DEK ERBB2 FGF21 GFRA4 HNF1B IL3RA JUP LYL1 ACTC1 BAG4 CAPRIN2 CDK3 CSNK1E DHFR ERBB3 FGF22 GGCX HNRNPA3 IL4R KAT2A LYN ACVR1 BAI3 CARD10 CDK4 CTCF DHH ERBB4 FGF23 GHR HOXA10 IL5RA KAT2B LZTR1 ACVR1B BAP1 CARD11 CDK5 CTCFL DIAPH1 ERCC1 FGF3 GID4 HOXA11 -

PRAK (MAPKAPK5) Antibody (T182) Peptide Affinity Purified Rabbit Polyclonal Antibody (Pab) Catalog # Ap7216a
9765 Clairemont Mesa Blvd, Suite C San Diego, CA 92124 Tel: 858.875.1900 Fax: 858.622.0609 PRAK (MAPKAPK5) Antibody (T182) Peptide Affinity Purified Rabbit Polyclonal Antibody (Pab) Catalog # AP7216a Specification PRAK (MAPKAPK5) Antibody (T182) - Product Information Application WB,E Primary Accession Q8IW41 Reactivity Human Host Rabbit Clonality Polyclonal Isotype Rabbit Ig Clone Names RB13233 Calculated MW 54220 Antigen Region 160-189 PRAK (MAPKAPK5) Antibody (T182) - Additional Information Gene ID 8550 Other Names MAP kinase-activated protein kinase 5, Western blot analysis of MAPKAPK5 Antibody MAPK-activated protein kinase 5, MAPKAP (T182) (Cat.#AP7216a) in Hela cell line lysates kinase 5, MAPKAP-K5, MAPKAPK-5, MK-5, (35ug/lane). MAPKAPK5 (arrow) was detected MK5, p38-regulated/activated protein kinase, using the purified Pab. PRAK, MAPKAPK5, PRAK Target/Specificity This PRAK(MAPKAPK5) antibody is generated from rabbits immunized with a KLH conjugated synthetic peptide between 160-189 amino acids from human PRAK(MAPKAPK5). Dilution WB~~1:1000 Format Purified polyclonal antibody supplied in PBS with 0.09% (W/V) sodium azide. This antibody is purified through a protein A column, followed by peptide affinity purification. Storage Maintain refrigerated at 2-8°C for up to 6 Western blot analysis of MAPKAPK5 (arrow) months. For long term storage store at -20°C using rabbit polyclonal MAPKAPK5 Antibody in small aliquots to prevent freeze-thaw (T182) (Cat.#AP7216a). 293 cell lysates (2 cycles. ug/lane) either nontransfected (Lane 1) or transiently transfected (Lane 2) with the Precautions MAPKAPK5 gene. PRAK (MAPKAPK5) Antibody (T182) is for research use only and not for use in diagnostic or therapeutic procedures. -

Significance of Serine Threonine Tyrosine Kinase 1 As a Drug Resistance Factor and Therapeutic Predictor in Acute Leukemia
INTERNATIONAL JOURNAL OF ONCOLOGY 45: 1867-1874, 2014 Significance of serine threonine tyrosine kinase 1 as a drug resistance factor and therapeutic predictor in acute leukemia Shinya Nirasawa, DAISUKE Kobayashi, TAKASHI KONDOH, KAGEAKI Kuribayashi, MAKI TANAKA, NOZOMI YANAGIHARA and NAOKI Watanabe Department of Clinical Laboratory Medicine, Sapporo Medical University School of Medicine, Sapporo 060-8543, Japan Received June 13, 2014; Accepted July 30, 2014 DOI: 10.3892/ijo.2014.2633 Abstract. Alterations in the mRNA expression or the tutively in patients before therapy and that promote natural mutation of previously reported tyrosine kinases have been resistance. detected only in a limited number of patients with acute The overexpression and mutation of various tyrosine leukemia. In this study, we examined whether the widely kinases contributes to the development of acute leukemia. For expressed serine threonine tyrosine kinase 1 (STYK1)/novel example, the overexpression or mutation of tyrosine kinases, oncogene with kinase domain (NOK) acts as a drug resis- including Flt3, c-kit, platelet-derived growth factor receptor tance factor in acute leukemia. The transfection of leukemic and Bcr-Abl has been reported (1-3). Once these tyrosine HL-60 cells with an STYK1 expression vector resulted in kinases are activated, they transmit major molecules such as the resistance to doxorubicin and etoposide and decreased phosphatidylinositol 3-kinase (PI3K) and mitogen-activated drug-induced caspase-3/7 activity and sub-G1 popula- protein kinase (MAPK) (1-3). This not only induces prolif- tion. To investigate the mechanism of STYK1-induced eration of leukemia cells, but also renders them resistant to drug resistance, microarray analysis was performed using various anticancer drugs.