ELLA ANLE KASANGA.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Lake Tanganyika, Regional Fisheries Programme (TREFIP)
FAO/NORWAY GOVERNMENT GCP/INT/648/NOR COOPERATIVE PROGRAMME Field Report F-14 (En) eries FISHCODE MANAGEMENT LAKE TANGANYIKA REGIONAL FISHERIES PROGRAMME (TREFIP) PREPARED BY THE JOINT AfDB/FAO/FISHCODE MISSION C. MAGNET, J.E. REYNOLDS AND H. BRU FOOD AND AGRICULTURE ORGANIZATION OF THE UNITED NATIONS ROME, JULY 2000 FAO/Norway Programme of Assistance to Developing Countries for the Implementation of the Code of Conduct for Responsible of the Code Conduct FAO/NorwayFish Programme of Assistance to Developing Countries for the Implementation Fisheries Management for the Provision Advice of Scientific for Improving Countries to Developing Assistance F: Sub-programme LAKE TANGANYIKA REGIONAL FISHERIES PROGRAMME (TREFIP) A proposal for implementation of the Lake Tanganyika Framework Fisheries Management Plan Prepared by: The Joint AfDB/FAO/FISHCODE Lake Tanganyika Mission Christophe Magnet (Team Leader/Economist, AfDB), J.Eric Reynolds (Development Planner/Socio-Economist, FAO), & Hervé Bru (Infrastructure/Marketing Specialist, AfDB) African Development Bank, Food and Agriculture Organization Abidjan of the United Nations, Rome July 2000 The designations employed and the presentation of material in this publication do not imply the expression of any opinion whatsoever on the part of the Food and Agriculture Organization of the United Nations concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. LAKE TANGANYIKA REGIONAL FISHERIES PROGRAMME (TREFIP) 18.07.00 ACKNOWLEDGEMENTS This document was drafted on behalf of the AfDB and the four Lake Tanganyika littoral States of Burundi, the Democratic Republic of Congo (DRC), Tanzania, and Zambia. Responsibility for its preparation was assigned to the Fisheries Policy and Planning Service (FIPP) of FAO, with funding provided by the AfDB and the FAO FISHCODE Programme (GCP/INT/648/NOR -- Interregional Programme of Assistance to Developing Countries for the Implementation of the Code of Conduct for Responsible Fisheries). -

WAI-ZEI Paper No. 21
Ludger Kühnhardt World War I: Lessons Learned and Lessons Threatened WAI-ZEI PAPER WAI-ZEI No. 21 2015 Professor Dr. Ludger Kühnhardt has been Director at the Center for European Integration Studies (ZEI) since October 1, 1997 and is in charge of ZEI‘s political and cultural research program. He is also a Professor at the Institute for Political Science and Sociology at the University of Bonn. Professor Kühnhardt‘s main research specializes in issues of the European Integration, the global comparison of regional integration systems as well as in topics that concern political theory and philosophy. Ludgern Kühnhardt studied history, political science and philosophy in Bonn, Geneva, Harvard and Tokyo. He received his PhD in 1983 and habilitated in 1986, both times specializing in political science at the University of Bonn. Between 1985 and 1987 Kühnhardt worked as a research assistant for Prof. Dr. Drs. h.c. Karl Dietrich Bracher at the Institute for Political Science at the University of Bonn. From 1987 to 1989 he worked as speechwriter for the President of the Federal Republic of Germany, Richard von Weizsäcker. Kühnhardt was chair for Political Science at the Albert-Ludwigs-University Freiburg between 1991 and 1997. In 1994/95 he served as Dean of the Philosophical Faculty at that University. He was Visiting Professor at the Universities of Jena, Capetown, at the College of Europe (Collège d’Europe) in Bruges, at the Alta Scuola de Economia e Relazioni Internazionali ASERI, at the Catholic University of Milan (since 1997), at Dartmouth College (New Hampshire), at the Diplomatic Academy Vienna (since 2002), at Stanford University, at Seoul National University, at the Diplomatic Academy of Mediterranean Studies (MEDAC), Malta (since 2007) and at St Antony’s College Oxford. -

Maritime Trade on Lake Tanganyika Trade Opportunities for Zambia
Maritime Trade on Lake Tanganyika Trade Opportunities for Zambia Commissioned by the Netherlands Enterprise Agency Maritime Trade on Lake Tanganyika Trade Opportunities for Zambia Maritime Trade on Lake Tanganyika Trade Opportunities for Zambia Rotterdam, July 2019 Table of contents Preface 3 Abbreviations and Acronyms 4 1 Introduction 5 2 Transport and Logistics 10 3 International and Regional Trade 19 4 Trade Opportunities 29 5 Recommendations and Action Plan 41 References 48 Annex A Trade Statistics 50 Annex B Trade Potential 52 Annex C Maps 53 Maritime Trade on Lake Tanganyika 2 Preface This market study was prepared by Ecorys for the Netherlands Enterprise Agency (RVO). The study provides information on trade opportunities between the countries on the shores of Lake Tanganyika, with a particular focus on Zambia and the port in Mpulungu. As such this study fills a gap, as previous studies were mostly focused on the infrastructure and logistics aspects of maritime trade on Lake Tanganyika. *** The study was prepared by Michael Fuenfzig (team leader & trade expert), Mutale Mangamu (national expert), Marten van den Bossche (maritime transport expert). We also thank Niza Juma from Ecorys Zambia (PMTC) for her support. This study is based on desk research, the analysis of trade statistics, and site visits and interviews with stakeholders around Lake Tanganyika. In Zambia Lusaka, Kasama, Mbala and Mpulungu were visited, in Tanzania, Kigoma and Dar es Salaam, and in Burundi, Bujumbura. The study team highly appreciates all the efforts made by the RVO, the Netherlands Ministry of Foreign Affairs and other stakeholders. Without their cooperation and valuable contributions this report would not have been possible. -

A Partner of Choice for the Eastern Africa We Want
THE AFRICAN DEVELOPMENT BANK A Partner of Choice for the Eastern Africa we want The African Development Bank A Partner of Choice for the Eastern Africa we want THE AFRICAN DEVELOPMENT BANK A PARTNER OF CHOICE FOR THE EASTERN AFRICA WE WANT THE AFRICAN DEVELOPMENT BANK GROUP This report has been prepared by the African Development Bank (AfDB) Group. Designations employed in this publication do not imply the expression of any opinion on the part of the institution concerning the legal status of any country, or the limitation of its frontier. While efforts have been made to present reliable information, the AfDB accepts no responsibility whatsoever for any consequences of its use. Vice President: Zondo Sakala Regional Director (EARC): Gabriel Negatu Lead Economist: Stefan Muller Regional Integration Specialist: Robert Rudy Consultants: Andy Dijkerman, James Adams Copyright 2014 — AFRICAN DEVELOPMENT BANK GROUP Photo Credits: AfDB photo fi les. PUBLISHED BY African Development Bank Group - East Africa Regional Resource Centre (EARC) Khushee Tower Longonot Road, Upper Hill Nairobi, Kenya Phone: (254) 20 2712925/26/28 Fax: (254) 20 2712938 Email: [email protected] Website: www.afdb.org 4 | AFRICAN DEVELOPMENT BANK GROUP Table of Contents List of Figures ii List of Tables iii List of Text Boxes iv List of Project Showcase Profi les iv Abbreviations and Acronyms v Acknowledgements vi Preface vii Executive Summary viii 1 Eastern Africa — A Region on the Rise Aided by the AfDB as a Partner of Choice 1 1.1 A Region on the Rise 2 1.2 The AfDB as a Partner -
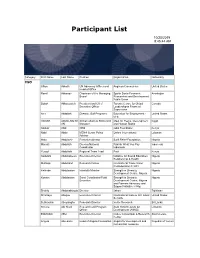
Participant List
Participant List 10/20/2019 8:45:44 AM Category First Name Last Name Position Organization Nationality CSO Jillian Abballe UN Advocacy Officer and Anglican Communion United States Head of Office Ramil Abbasov Chariman of the Managing Spektr Socio-Economic Azerbaijan Board Researches and Development Public Union Babak Abbaszadeh President and Chief Toronto Centre for Global Canada Executive Officer Leadership in Financial Supervision Amr Abdallah Director, Gulf Programs Educaiton for Employment - United States EFE HAGAR ABDELRAHM African affairs & SDGs Unit Maat for Peace, Development Egypt AN Manager and Human Rights Abukar Abdi CEO Juba Foundation Kenya Nabil Abdo MENA Senior Policy Oxfam International Lebanon Advisor Mala Abdulaziz Executive director Swift Relief Foundation Nigeria Maryati Abdullah Director/National Publish What You Pay Indonesia Coordinator Indonesia Yussuf Abdullahi Regional Team Lead Pact Kenya Abdulahi Abdulraheem Executive Director Initiative for Sound Education Nigeria Relationship & Health Muttaqa Abdulra'uf Research Fellow International Trade Union Nigeria Confederation (ITUC) Kehinde Abdulsalam Interfaith Minister Strength in Diversity Nigeria Development Centre, Nigeria Kassim Abdulsalam Zonal Coordinator/Field Strength in Diversity Nigeria Executive Development Centre, Nigeria and Farmers Advocacy and Support Initiative in Nig Shahlo Abdunabizoda Director Jahon Tajikistan Shontaye Abegaz Executive Director International Insitute for Human United States Security Subhashini Abeysinghe Research Director Verite -

Western Tanzania Is Rough, Remote Frontier Land, with Vast Tabora
©Lonely Planet Publications Pty Ltd We s t e r n T a n z a n i a Why Go? Western Tanzania is rough, remote frontier land, with vast Tabora ...........................230 trackless expanses, minimal infrastructure and very few vis- Kigoma .........................232 itors: much as it was back when Stanley found Livingstone Ujiji ................................235 here. The west serves a sense of adventure now extinct in Gombe National Park .....236 the rest of the country; and this is precisely what attracts Mahale Mountains a trickle of travellers, many of whom plan their itineraries National Park................238 around the schedules of the MV Liemba, which sails down Uvinza ...........................240 Lake Tanganyika and the Central Line train, which crosses Mpanda.........................240 the country. Katavi National Park ....242 But, it’s wildlife watching that brings most people. Go- Sumbawanga................244 mbe, Jane Goodall’s former stomping grounds, and Mahale Mountains National Parks are two of the world’s best places for chimpanzee encounters, while the vast fl oodplains of rarely visited Katavi National Park off er an almost primeval Best of Culture safari experience. » MV Liemba (p 374 ) Unless you use chartered planes as part of a tour, you’ll need plenty of time and even more patience to travel here. » Katonga (p 232 ) But, for that certain sort of traveller, Tanzania’s west is » Kipili (p 240 ) Tanzania’s best. » Livingstone’s tembe (p 230 ) When to Go Kigoma Best of Nature °C/°F Temp Rainfall inches/mm 40/104 16/400 » Mahale Mountains National Park (p 238 ) 30/86 12/300 » Katavi National Park 20/68 8/200 (p 242 ) » Gombe National Park 10/50 4/100 (p 236 ) 0/32 0 » Lake Tanganyika diving J FDNOSAJJMAM and snorkelling (p 240) Dec-Apr Rains May-Nov Dry- May-Jun Chim- » Kalambo Falls (p 244 ) bring washed-out season travel panzees are most roads but brilliant is easiest, but likely to be seen lightning displays. -

The Hydrology of Lake Tanganyika
Bulletin No. 6. TANGANYIKA TERRITOR Y GEOLOGICAL SURVEY DEPARTMENT The Hydrology of Lake Tanganyika By C. GILLMAN, F.G.S., Chief Engineer, Tanganyika Railways and Ports Services PREFACE. 'L'he subject of the hydrology of the lakes of Equatorial Africa is one of outstanding geographical importance in many ways. This contribution regarding Lake Tanganyika is a welcome and valuable one to both the scientific and economic aspects of African geography. The subject is on the borderline of geological research, one aspect of which in this country is concerned with various phases in the history of the ancient lake systems and their deposits. A consideration of existing conditions is therefore a necessary stcp towards the satisfactory intel'pretation of earlier cycles. The geographical aspects of this subject have a definite geological relationship and it is hoped that in due course a review of the fascinating subject of the old lake systems and thcir deposits will follow as a sequel to the present paper. E. O. TEALE, Director. CONTENTS. PAGE. I.-INTRODUCTORY I n.-SUBDIVISIONS 0]' 'l'HE BASIN IlL-CLIMATIC CONSIDERA1'IONS: 3 Rainfall Evaporation IV.-CJ;IARACTERIS'l'ICS OF THE W A'fER: 3 Temperature Chemical Composition Hydraulics V.-DEP'l'H OF THE LAKE 4 VI,-FLUCTUA'l'IONS OF LAKE LEVEL: 5 Lukuga outlet Discharge Long amplitude Periodic Fluctuations Short amplitude Periodic Fluctuations Annual Fluctuations Daily Fluctuations VIl.-THE REGIME OF THE LAKE 10 VIlI.-FuTURE CHANGES AND TECHNICAL CONSIDERATIONS II IX.-COMPARISON Wll'H O1'HER CEN'mAL AFRICAN LAKES 13 X.-CORRELATION WI'l'H SOLAR ACnVITY .. -

A Checklist of the Land Mammals Tanganyika Territory Zanzibar
274 G. H. SWYNNERTON,F.Z.S., Checklist oj Land Mammals VOL. XX A Checklist of the Land Mammals OF mE Tanganyika Territory AND mE Zanzibar Protectorate By G. H. SWYNNERTON, F.Z.S., Game Warde:z, Game Preservation Department, Tanganyika Territory, and R. W. HAYMAN, F.Z.S., Senior Experimental Officer, Department of Zoology, British Museum (Natural History) 277278·.25111917122896 .· · 4 . (1)(3)(-)(2)(5)(9)(3)(4)280290281283286289295288291 280. .. CONTENTS· · · No. OF FORMS* 1. FOREWORDINSECTIVORA ErinaceidaM:,gadermatidaEmballonuridaSoricidt:eMacroscelididaMarossidaNycteridaHipposideridaRhinolophidaVespertilionida(Shrews)(Free-tailed(Hollow-faced(Hedgehogs)(Horseshoe(Leaf-nosed(Sheath-tailed(Elephant(Simple-nosed(Big-earedBats)Bats)Shrews)BatsBats)Bats) Pteropodida (Fruit-eating Bats) 2.3. INTRODUCTIONSYSTEMATICLIST OF SPECIESAND SUBSPECIES: PAGE CHIROPTERA Chrysochlorida (Golden" Moles to) ···302306191210.3521. ·2387 . · 6 · IAN. (1)(2)1951(-)(4)(21)(1)(6)(14)(6)(5),(7)(8)333310302304306332298305309303297337324325336337339211327 . SWYNNERTON,. P.Z.S.,·· ·Checklist··· of·Land 3293Mammals52 275 PItIMATES G. It. RhinocerotidaPelidaEchimyidaHyanidaPongidaCercopithecidaHystricidaMuridaHominidaAnomaluridaPedetidaCaviidaMustelidaGliridaSciuridaViverrida(Cats,(Mice,(Dormice)(Guinea-pigs)(Apes)(Squirrels)(Spring(Hyaenas,(Genets,(Man)(Polecats,(Cane(porcupines)(Flying(Rhinoceroses)Leopards,(Monkeys,Rats,Haas)Rats)Civets,Arad-wolf).Weasels,Squirrels)Gerbils,Lions,Baboons)Mongooses)Ratels,etc.)•Cheetahs)..Otters) ProcaviidaCanidaLeporidaElephantidaLorisidaOrycteropodidaEquidaBathyergidaManida -

"German" East Africa Author(S): Owen Letcher Source: the Geographical Journal, Vol
Notes on the South-Western Area of "German" East Africa Author(s): Owen Letcher Source: The Geographical Journal, Vol. 51, No. 3 (Mar., 1918), pp. 164-172 Published by: geographicalj Stable URL: http://www.jstor.org/stable/1779377 Accessed: 23-06-2016 09:06 UTC Your use of the JSTOR archive indicates your acceptance of the Terms & Conditions of Use, available at http://about.jstor.org/terms JSTOR is a not-for-profit service that helps scholars, researchers, and students discover, use, and build upon a wide range of content in a trusted digital archive. We use information technology and tools to increase productivity and facilitate new forms of scholarship. For more information about JSTOR, please contact [email protected]. Wiley, The Royal Geographical Society (with the Institute of British Geographers) are collaborating with JSTOR to digitize, preserve and extend access to The Geographical Journal This content downloaded from 132.236.27.111 on Thu, 23 Jun 2016 09:06:43 UTC All use subject to http://about.jstor.org/terms 164 NOTES ON THE the main trunk railway through Siberia, and it naturally makes it impossible without very large capital to develop these resources. But the policy of the Government before the revolution was to develop rail way s as rapidly as pos? sible ; even now construction is going on in many parts of that country; and there is every probability that after the war the attention of miners all over the world will be attracted to Siberia. The political outlook at the present moment can be regarded as only a passing phase. -

The Study of Master Plan for Port Sector in the Republic of Burundi
Ministry of Transport, Public Works and Equipment Burundi The Study of Master Plan for Port Sector in the Republic of Burundi Final Report September 2012 JAPAN INTERNATIONAL COOPERATION AGENCY PADECO Co., Ltd. Nippon Koei Co. Ltd. International Development Center of Japan Incorporated EI JR 12-189 Ministry of Transport, Public Works and Equipment Burundi The Study of Master Plan for Port Sector in the Republic of Burundi Final Report September 2012 JAPAN INTERNATIONAL COOPERATION AGENCY PADECO Co. Ltd. Nippon Koei Co. Ltd. International Development Center of Japan Incorporated The Study of Master Plan for Port Sector in the Republic of Burundi Final Report Contents Abbreviations and Acronyms .................................................................................................. viii Chapter 1 Introduction .................................................................................................... 1-1 1.1 Background of the Study ........................................................................................... 1-1 1.2 Objectives of the Study .............................................................................................. 1-2 1.3 Study Area ................................................................................................................. 1-2 Chapter 2 Socio/Economic Developments in Burundi .................................................. 2-1 2.1 Trends in Socio/Economic Status .............................................................................. 2-1 2.1.1 General Trend .............................................................................................. -

Western Tanzania
© Lonely Planet Publications 257 Western Tanzania The west is Tanzania’s rough, remote frontier land, with few tourists, minimal infrastructure, vast trackless expanses crossed only by the ageing Central Line train and little to draw you here – unless you’re interested in chimpanzees. For this, and for watching wildlife in one of WESTERN TANZANIA Tanzania’s most pristine settings, it’s a fascinating destination. Gombe Stream National Park – Jane Goodall’s world-renowned chimpanzee research sta- tion – and Mahale Mountains National Park offer excellent opportunities to get close to our primate cousins. At Katavi National Park, you’ll be just a speck in the surrounding universe of vast floodplains trammelled by thousands of buffaloes, plus zebras, lions and more. Those with a sense of adventure and imagination can visit tiny Ujiji. Now it’s a nondescript fishing village, but in its heyday it was the terminus of one of East Africa’s most important caravan routes, linking Lake Tanganyika with Bagamoyo and the sea, an important dhow-building centre and a way station for several European expeditions. Lake Tanganyika itself is a scenic and useful transport route if you are heading to or from northern Zambia, and makes a welcome respite from dusty, bumpy roads, with some unforgettable sunset views. Wherever you go, travel in western Tanzania is rugged, and you will need plenty of time. There are few roads (none of them good), and often the only transport choices are boat, train or truck. Outside of Kigoma, Tabora and the national parks, the region is seldom visited and has few facilities. -

Tanzania | a Leader Among Africa’S Emerging Markets
Tanzania | A Leader among Africa’s Emerging Markets Tanzania A Leader among Africa’s Emerging Markets October 2016 01 Tanzania | A Leader among Africa’s Emerging Markets Brief overview The United Republic of Tanzania (Tanzania) has recorded an annual average growth rate of more than 6% over the past decade and is on course to maintain a robust growth rate of over 6.5% going forward. Despite global economic and financial uncertainties, the economy has been able to achieve these consistent growth rates coupled with a low inflation rate, driven by activity in sectors such as mining, energy, construction and manufacturing. Improving public sector efficiency and a crackdown on corruption has been the focus of the new administration under the leadership of President John Magufuli, elected in 2015. The government intends to stimulate inclusive growth and reduce poverty levels by running a leaner administration, promoting tax compliance, building Private-Public Partnerships (PPPs) and attracting investment into industrial sector development. Underpinned by favourable demographics and supported by a government that is showing signs of principled leadership with intentions to invest in education, skills transfers and infrastructure to drive growth, Tanzania is well-positioned to continue on its current rapid growth path. Its young and culturally-diverse population of more than 50 million makes it eastern Africa’s second most- populous nation after Ethiopia; expected to reach almost 83 million by 2030. Greater emphasis on upscaling urban hard and soft infrastructure and creating employment opportunities in light of a rapidly-growing urban population will be integral in supporting its national development vision, the Tanzania Development Vision (TDV) that looks to transform the economy into a middle-income and semi-industrialised state by 2025.