Australian Public Assessment Report Ranolazine
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Drug Class Review Beta Adrenergic Blockers
Drug Class Review Beta Adrenergic Blockers Final Report Update 4 July 2009 Update 3: September 2007 Update 2: May 2005 Update 1: September 2004 Original Report: September 2003 The literature on this topic is scanned periodically. The purpose of this report is to make available information regarding the comparative effectiveness and safety profiles of different drugs within pharmaceutical classes. Reports are not usage guidelines, nor should they be read as an endorsement of, or recommendation for, any particular drug, use, or approach. Oregon Health & Science University does not recommend or endorse any guideline or recommendation developed by users of these reports. Mark Helfand, MD, MPH Kim Peterson, MS Vivian Christensen, PhD Tracy Dana, MLS Sujata Thakurta, MPA:HA Drug Effectiveness Review Project Marian McDonagh, PharmD, Principal Investigator Oregon Evidence-based Practice Center Mark Helfand, MD, MPH, Director Oregon Health & Science University Copyright © 2009 by Oregon Health & Science University Portland, Oregon 97239. All rights reserved. Final Report Update 4 Drug Effectiveness Review Project TABLE OF CONTENTS INTRODUCTION .......................................................................................................................... 6 Purpose and Limitations of Evidence Reports........................................................................................ 8 Scope and Key Questions .................................................................................................................... 10 METHODS................................................................................................................................. -

Inhibitory Effect of Eslicarbazepine Acetate and S-Licarbazepine on 2 Nav1.5 Channels
bioRxiv preprint doi: https://doi.org/10.1101/2020.04.24.059188; this version posted August 14, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 Inhibitory effect of eslicarbazepine acetate and S-licarbazepine on 2 Nav1.5 channels 3 Theresa K. Leslie1, Lotte Brückner 1, Sangeeta Chawla1,2, William J. Brackenbury1,2* 4 1Department of Biology, University of York, Heslington, York, YO10 5DD, UK 5 2York Biomedical Research Institute, University of York, Heslington, York, YO10 5DD, UK 6 * Correspondence: Dr William J. Brackenbury, Department of Biology and York Biomedical 7 Research Institute, University of York, Wentworth Way, Heslington, York YO10 5DD, UK. Email: 8 [email protected]. Tel: +44 1904 328284. 9 Keywords: Anticonvulsant, cancer, epilepsy, eslicarbazepine acetate, Nav1.5, S-licarbazepine, 10 voltage-gated Na+ channel. 11 Abstract 12 Eslicarbazepine acetate (ESL) is a dibenzazepine anticonvulsant approved as adjunctive treatment for 13 partial-onset epileptic seizures. Following first pass hydrolysis of ESL, S-licarbazepine (S-Lic) 14 represents around 95 % of circulating active metabolites. S-Lic is the main enantiomer responsible 15 for anticonvulsant activity and this is proposed to be through the blockade of voltage-gated Na+ 16 channels (VGSCs). ESL and S-Lic both have a voltage-dependent inhibitory effect on the Na+ current 17 in N1E-115 neuroblastoma cells expressing neuronal VGSC subtypes including Nav1.1, Nav1.2, 18 Nav1.3, Nav1.6 and Nav1.7. -

Formulation and Evaluation of Ranolazine Extended Release Tablets: Influence of Polymers
Formulation and evaluation of ranolazine extended release tablets: Influence of polymers Murthy TEGK, Bala Vishnu Priya Mukkala, Suresh Babu VV1 Department of Pharmaceutics, Bapatla College of Pharmacy, Bapatla, Guntur, Andhra Pradesh, 1Natco Pharma Limited, Kothur, Hyderabad, Andhra Pradesh, India n extended release tablet provides prolonged periods of drug in plasma levels thereby reduce dosing frequency, Aimprove patient compliance and reduce the dose-related side effects. Ranolazine is indicated for the chronic treatment of angina in patients who have not achieved an adequate response with other anti-anginal agents. The present investigation was undertaken to design extended release tablets of Ranolazine employing hypromellose phthalate grade HP-55, ethocel standard 7FP premium ethyl cellulose, Surelease E-7-19040, Klucel HF pharm and Natrosol Type 250 HHX as matrix forming RESEARCH ARTICLE agents using wet granulation method. Formulated tablets were evaluated for uniformity of weight, assay, water content, in vitro drug release studies and stability studies. The drug release followed first order kinetics with both erosion and diffusion as the release mechanism. It is concluded that the desired drug release pattern can be obtained by using natrosol type 250 HHX compared to other polymers. The similarity factor (f2) was calculated to select best formulation by comparing in vitro dissolution data of the commercial formulation Ranexa®. The formulated tablets fulfilled the compendia requirements. The formulated Ranolazine Extended release tablets were found to be stable. Key words: Ehocel, extended release tablets, hypromellose phthalate, klucel HF pharm and natrosol, ranolazine, surelease INTRODUCTION MATERIALS AND METHODS Extended release drug delivery technology can provide Ranolazine was procured from the Natco Pharma smooth plasma levels of drug over longer periods of Ltd, Hyderabad. -

Ranexa, INN:Ranolazine
European Medicines Agency Doc. Ref.: EMEA/CHMP/643056/2009 EMEA/H/C/805 Ranexa1 ranolazine EPAR summary for the public This document is a summary of the European Public Assessment Report (EPAR). It explains how the Committee for Medicinal Products for Human Use (CHMP) assessed the studies performed, to reach their recommendations on how to use the medicine. If you need more information about your medical condition or your treatment, read the Package Leaflet (also part of the EPAR) or contact your doctor or pharmacist. If you want more information on the basis of the CHMP recommendations, read the Scientific Discussion (also part of the EPAR). What is Ranexa? Ranexa is a medicine that contains the active substance ranolazine. It is available as oval prolonged- release tablets (blue: 375 mg; orange: 500 mg; green: 750 mg). ‘Prolonged release’ means that ranolazine is released slowly from the tablet over a few hours. What is Ranexa used for? Ranexa is used to treat the symptoms of stable angina pectoris (chest pain caused by reduced blood flow to the heart). It is used as an add-on to existing treatment in patients whose disease is not adequately controlled by other medicines for angina pectoris, such as beta-blockers or calcium antagonists, or in patients who cannot take these medicines. The medicine can only be obtained with a prescription. How is Ranexa used? The recommended starting dose of Ranexa is 375 mg twice a day. After two to four weeks, the dose should be increased to 500 mg twice a day, and then to 750 mg twice a day, depending on the patient’s response. -

Medcompgx Med List
For more info, go to: MedCompGx.com Medications Conditions / Uses Other Uses CYP2C19 Anticonvulsants Lacosamide (Vimpat) Epilepsy, Seizures Phenobarbital (Luminal) Epilepsy, Seizures Barbituates, Anxiety, Sedation, Withdrawl Primidone (Mysoline) Epilepsy, Seizures Zonisamide (Zonegran) Epilepsy, Seizures Antifungals Voriconazole (Vfend) Yeast Infections, Candidiasis, Aspergillosis, Candida, Aspergillus Antidepressants Escitalopram (Lexapro) SSRI, Depression, Anxiety Citalopram (Celexa) SSRI, Depression, Anxiety OCD Sertraline (Zoloft) SSRI, Depression, Anxiety OCD, PTSD Doxepin (Silenor) Tricyclic Antidepressant, Depression Bipolar, Insomnia Imipramine (Tofranil) Tricyclic Antidepressant, Depression Trimipramine (Surmontil) Tricyclic Antidepressant, Depression Amitriptyline (Elavil) Tricyclic Antidepressant, Depression Clomipramine (Anafranil) Tricyclic Antidepressant, Depression OCD Benzodiazepines Clobazam (Onfi) Seizures Diazepam (Valium) Seizures, Muscle Spasms, Anxiety, Alcohol Withdrawl Beta Blockers Labetalol (Normodyne, Trandate) High Blood Pressure, Hypertension Immunomodulators Leflunomide (Arava) Rheumatoid Arthritis Tofacitinib (Xeljanz) Rheumatoid Arthritis Muscle Relaxants Carisoprodol (Soma) Muscle Pain, Muscle Spasms Opioids Meperidine (Demerol) Pain, Severe Pain, Narcotic, Opiate Proton Pump Inhibitors Dexlansoprazole (Dexilant) GERD, Acid Reflux, Gastric Esophageal Reflux, Ulcers, Heartburn, Zollinger-Ellison Syndrome Esomeprazole (Nexium) GERD, Acid Reflux, Gastric Esophageal Reflux, Ulcers, Heartburn, Zollinger-Ellison -

Ranolazine Doesn't Need BP Reduction to Work
March 2007 • www.ecardiologynews.com Acute Coronary Syndromes 17 Ranolazine Doesn’t Need BP Reduction to Work BY BETSY BATES thought to block the late sodium current lates into important clinical information, severe renal impairment, ranolazine did in- Los Angeles Bureau that results from a decreased oxygen sup- because its antianginal and anti-ischemic crease blood pressure by 10-15 mm Hg, so ply to the heart, thereby reducing the effects do not depend on reductions in “you need to be careful” and monitor S NOWMASS, COLO. — A year-old drug sodium-dependent calcium overload that heart rate or blood pressure. blood pressure regularly. believed to inhibit ischemia through a nov- acts as a key player in the development of “All of the other drugs do,” said Dr. Dr. Conti said the “important trial that el mechanism may prove useful for angina ischemia, explained Dr. Conti at the meet- Conti, eminent scholar and professor of everyone knows about” was the Combi- patients whose symptoms are not relieved ing, which was cosponsored by the Amer- cardiology at the University of Florida, nation Assessment of Ranolazine in Stable by revascularization or other agents, Dr. C. ican Academy of Cardiology. Gainesville. Angina (CARISA), in which ranolazine Richard Conti said at a conference spon- It is the first major advance in antian- In clinical trials, patients experienced significantly improved exercise duration sored by the Society for Cardiovascular ginal therapy since calcium channel block- minimal changes in mean heart rate (less and angina onset at trough and peak dose, Angiography and Interventions. ers, he noted. -

Anaesthetic Guideline for the Management of Children with Long QT Syndrome SOP/Protocol Detail Owner: Dr
Anaesthetic Guideline for the management of children with long QT Syndrome SOP/Protocol Detail Owner: Dr. Jutta Scheffczik Publication: December 2020 Review: December 2023 Aims To ensure the safety of paediatric patients with congenital or acquired long QT syndrome who need a general anaesthetic. It is anticipated that most patients will undergo anaesthesia in Leeds and this guideline is to support those patients who do need to have a general anaesthetic at their local hospital. Objectives 1.To provide guidance on the management of children with LQTS 2.To enable children to have minor surgery in their local hospital where appropriate 3.To improve equity and consistency in care across the Yorkshire and Humber CHD Network Background Long QT syndrome is a congenital or acquired channelopathy, impairing myocardial electrical conduction that results in impaired ventricular repolarization and can present clinically as recurrent syncope, pseudo-seizures, or sudden death. Patients with QT prolongation and LQTS are susceptible to the development of the characteristic polymorphic ventricular tachycardia, called Torsades de Pointes TdP. The prolongation of the QT interval caused by anaesthetic drugs and the sympathetic response to anaesthesia and surgery can trigger malignant arrhythmias in patients with long QT syndrome. Patients with a genetic predisposition to LQTS may be asymptomatic and may have a normal resting QTc interval, it is possible for an episode of torsade de pointes to occur for the first time during anaesthesia. Diagnosis The diagnosis of long QT syndrome should be made by a paediatrician, a paediatrician with an expertise in cardiology or a paediatric cardiologist. QT intervals need to be corrected for heart rate (QTc – routinely defined using the Bazzett formula) and are highly variable, but the abnormal corrected values are defined as a pre-puberty average of >470ms in males, >480ms in females. -

Attachment: Product Information: Ranolazine
Attachment 1: Product information for AusPAR - AusPAR RANEXA ranolazine – A Menarini Australia Pty Ltd PM-2015-00423-1-3 FINAL 20 February 2018 This Product information was approved at the time this AusPAR was published. PRODUCT INFORMATION RANEXA® MODIFIED-RELEASE TABLETS NAME OF THE MEDICINE Ranolazine Chemical Structure Ranolazine is designated chemically as (±)-N-(2,6-dimethylphenyl)4-[2-hydroxy-3-(2- methoxyphenoxy)propyl] piperazineacetamide. The empirical formula for ranolazine is C24H33N3O4 with a molecular weight of 427.54. CAS number 95635-55-5 DESCRIPTION Ranolazine is a white to off-white solid powder. Ranolazine is soluble in dichloromethane and methanol; sparingly soluble in tetrahydrofuran, ethanol, acetonitrile, and acetone; slightly soluble in ethyl acetate, isopropanol, toluene, and ethyl ether; and very slightly soluble in water. Ranolazine is very slightly soluble at pH above 6.99, slightly soluble at pH from 6.29 to 5.76, sparingly soluble at pH 5.25, soluble at pH from 5.01 to 4.82 and freely soluble below pH 4.40. The partition coefficient was determined using three different ratios of octanol: water. The log P for ranolazine was measured at 2.07 ± 0.06. RANEXA 375 mg, 500 mg and 750 mg tablets contain the following inactive ingredients: carnauba wax, hypromellose, magnesium stearate, methacrylic acid-ethyl acrylate copolymer (1:1), microcrystalline cellulose, sodium hydroxide and titanium dioxide. Additional inactive ingredients for the 375 mg tablet include: macrogol, polysorbate 80, indigo carmine aluminium lake. Additional inactive ingredients for the 500 mg tablet include: macrogol, polyvinyl alcohol, iron oxide yellow, iron oxide red, purified talc. -

Federal Register / Vol. 60, No. 80 / Wednesday, April 26, 1995 / Notices DIX to the HTSUS—Continued
20558 Federal Register / Vol. 60, No. 80 / Wednesday, April 26, 1995 / Notices DEPARMENT OF THE TREASURY Services, U.S. Customs Service, 1301 TABLE 1.ÐPHARMACEUTICAL APPEN- Constitution Avenue NW, Washington, DIX TO THE HTSUSÐContinued Customs Service D.C. 20229 at (202) 927±1060. CAS No. Pharmaceutical [T.D. 95±33] Dated: April 14, 1995. 52±78±8 ..................... NORETHANDROLONE. A. W. Tennant, 52±86±8 ..................... HALOPERIDOL. Pharmaceutical Tables 1 and 3 of the Director, Office of Laboratories and Scientific 52±88±0 ..................... ATROPINE METHONITRATE. HTSUS 52±90±4 ..................... CYSTEINE. Services. 53±03±2 ..................... PREDNISONE. 53±06±5 ..................... CORTISONE. AGENCY: Customs Service, Department TABLE 1.ÐPHARMACEUTICAL 53±10±1 ..................... HYDROXYDIONE SODIUM SUCCI- of the Treasury. NATE. APPENDIX TO THE HTSUS 53±16±7 ..................... ESTRONE. ACTION: Listing of the products found in 53±18±9 ..................... BIETASERPINE. Table 1 and Table 3 of the CAS No. Pharmaceutical 53±19±0 ..................... MITOTANE. 53±31±6 ..................... MEDIBAZINE. Pharmaceutical Appendix to the N/A ............................. ACTAGARDIN. 53±33±8 ..................... PARAMETHASONE. Harmonized Tariff Schedule of the N/A ............................. ARDACIN. 53±34±9 ..................... FLUPREDNISOLONE. N/A ............................. BICIROMAB. 53±39±4 ..................... OXANDROLONE. United States of America in Chemical N/A ............................. CELUCLORAL. 53±43±0 -

(12) Patent Application Publication (10) Pub. No.: US 2008/0312247 A1 Gant Et Al
US 2008.0312247A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2008/0312247 A1 Gant et al. (43) Pub. Date: Dec. 18, 2008 (54) SUBSTITUTED PIPERAZINES Publication Classification (51) Int. Cl. (75) Inventors: Thomas G. Gant, Carlsbad, CA A63/495 (2006.01) (US); Sepehr Sarshar, Cardiff by A6IP 9/00 (2006.01) the Sea, CA (US) A6IP 9/10 (2006.01) C07D 24I/04 (2006.01) Correspondence Address: (52) U.S. Cl. .................................... 514/252.12:544/400 GLOBAL PATENT GROUP - APX (57) ABSTRACT Ms. LaVern Hall Disclosed herein are substituted piperazine late Na" channel 10411 Clayton Road, Suite 304 modulators of Formula I, process of preparation thereof, ST. LOUIS, MO 63131 (US) pharmaceutical compositions thereof, and methods of use thereof. (73) Assignee: AUSPEX PHARMACEUTICALS, INC., Formula I Vista, CA (US) Rs R14 Ris R10 R R31 (21) Appl. No.: 12/138,169 R6 R4 R11 R 23. R24V > R9 N N R O R56 N (22) Filed: Jun. 12, 2008 R O 2K O R30 R 8 R2 R ". 20R,\ R. R. Related U.S. Application Data R. R3 19 R22 R4 R29 (60) Provisional application No. 60/943,731, filed on Jun. R28 13, 2007. US 2008/0312247 A1 Dec. 18, 2008 SUBSTITUTED PPERAZINES nal of Clinical Pharmacology 2005, 45,802-09; Chaitman et al, Journal of the Americal College of Cardiology 2004, 43(8), 1375-82; Opie, European Heart Journal 2003, 24, 0001. This application claims the benefit of priority of 1854-56: Anderson et al, Heart Disease 2001, 3, 263-69; U.S. provisional application No. -
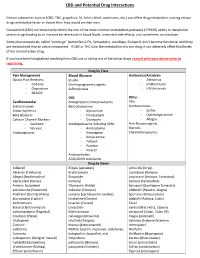
CBD and Potential Drug Interactions
CBD and Potential Drug Interactions Certain substances such as (CBD, THC, grapefruit, St. John’s Wort, watercress, etc.) can affect drug metabolism, making certain drugs metabolize faster or slower than they would on their own. Cannabidiol (CBD) can temporarily inhibit the one of the most common metabolism pathways (CYP450) ability to metabolize certain drugs leading to an increase (or decrease) in blood levels, unwanted side effects, and sometimes, an overdose. Some pharmaceuticals, called “prodrugs” (tamoxifen,5-FU, Simvastatin, Levodopa, Enalapril) don’t become functional until they are metabolized into an active component. If CBD or THC slow the metabolism of a pro-drug, it can adversely affect final levels of the desired active drug. If you have been hospitalized resulting from CBD use or taking any of the below drugs consult with your doctor prior to registering. Drug by Class Pain Management Blood Glucose Antibiotics/Antivirals Opioid Pain Relievers Insulin - Zithromax - Codeine Oral hypoglycemic agents - Erythromycin - Oxycodone Sulfonylureas - HIV Antivirals - NSAIDS CNS Other Cardiovascular Antiepileptics/anticonvulsants PPIs Statins (some) Benzodiazepines Antihistamines Antiarrhythmics - Alprazolam - Zyrtec Beta Blockers - Clonazepam - Diphenhydramine Calcium Channel Blockers - Diazepam - Allegra - Cardizem Antidepressants including SSRIs Anti-Nausea agents - Norvasc - Amitriptyline Steroids Anticoagulants - Amoxapine Chemotherapeutics - Desipramine - Tofranil - Pamelor - Vivactil Antipsychotics ADD/ADHD stimulants Drug by -

Brand Name: Aptiom Generic Name: Eslicarbazepine Manufacturer: Sunovion Pharmaceuticals Inc1 Product Availability: FDA Approved November 2013
Brand Name: Aptiom Generic Name: eslicarbazepine Manufacturer: Sunovion Pharmaceuticals Inc1 Product Availability: FDA approved November 2013. Anticipated availability is late 2014.2 Drug Class: Anticonvulsant, central nervous system agent, voltage-gated sodium channel blocker 3,4 Uses: Labeled uses: Adjunctive treatment of partial seizures3,4 Unlabeled uses: None o Under investigation for use as monotherapy for treatment of focal epilepsy or partial-onset seizures5 o Under investigation as a possible treatment for bipolar disorder3,5,6 Mechanism of Action: Like many other anticonvulsants, the exact mechanism of action of eslicarbazepine is unknown. The antiepileptic action of the drug is theorized to be due to the blockage of voltage-gated sodium channels. Studies indicate that eslicarbazepine and its metabolites competitively interact with voltage-gated sodium channels to inhibit sustained repetitive neuronal firing, with enhanced inhibitory selectivity for rapidly firing neurons.1,3 Pharmacokinetics: Absorption: 1,3 Tmax 1-4 hours Vd 61 L t ½ 13-20 hours Clearance 20 mL/min GFR 80-120 mL/min Protein binding < 40% (% to albumin unknown) Bioavailability > 90% Metabolism: 1,3 Eslicarbazepine is rapidly metabolized to its major active metabolite eslicarbazepine (91%) by hydrolytic first-pass metabolism. Minor active metabolites are R-licarbazepine (5%) and oxcarbazepine (1%). In in vitro studies, eslicarbazepine acetate was found to be a moderate inhibitor of CYP2C19 and an inducer of CYP3A4. Studies with eslicarbazepine in fresh human hepatocytes showed only mild activation of UGT1A1- mediated glucuronidation. Eslicarbazepine showed no clinical inhibitory effects on CYP1A2, CYP2A6, CYP2B6, CYP2D6, CYP2E1, and CYP3A4 and did not undergo autoinduction. Elimination: 1,3 Eslicarbazepine metabolites are eliminated primarily by renal excretion in the unchanged and glucuronide conjugate forms.