Summary of Product Characteristics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Inhaled Steroid Ciclesonide Blocks SARS-Cov-2 RNA Replication by Targeting Viral
bioRxiv preprint doi: https://doi.org/10.1101/2020.08.22.258459; this version posted August 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 The inhaled steroid ciclesonide blocks SARS-CoV-2 RNA replication by targeting viral 2 replication-transcription complex in culture cells 3 4 Shutoku Matsuyamaa#, Miyuki Kawasea, Naganori Naoa, Kazuya Shiratoa, Makoto Ujikeb, Wataru 5 Kamitanic, Masayuki Shimojimad, and Shuetsu Fukushid 6 7 aDepartment of Virology III, National Institute of Infectious Diseases, Tokyo, Japan 8 bFaculty of Veterinary Medicine, Nippon Veterinary and Life Science University, Tokyo, Japan 9 cDepartment of Infectious Diseases and Host Defense, Gunma University Graduate School of 10 Medicine, Gunma, Japan 11 dDepartment of Virology I, National Institute of Infectious Diseases, Tokyo, Japan. 12 13 Running Head: Ciclesonide blocks SARS-CoV-2 replication 14 15 #Address correspondence to Shutoku Matsuyama, [email protected] 16 17 Word count: Abstract 149, Text 3,016 bioRxiv preprint doi: https://doi.org/10.1101/2020.08.22.258459; this version posted August 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 18 Abstract 19 We screened steroid compounds to obtain a drug expected to block host inflammatory responses and 20 MERS-CoV replication. Ciclesonide, an inhaled corticosteroid, suppressed replication of MERS-CoV 21 and other coronaviruses, including SARS-CoV-2, the cause of COVID-19, in cultured cells. The 22 effective concentration (EC90) of ciclesonide for SARS-CoV-2 in differentiated human bronchial 23 tracheal epithelial cells was 0.55 μM. -

Genomic and Non-Genomic Effects of Aldosterone
132 Current Signal Transduction Therapy, 2012, 7, 132-141 Genomic and Non-genomic Effects of Aldosterone Andrea Verhovez*, Tracy A. Williams, Silvia Monticone, Valentina Crudo, Jacopo Burrello, Maddalena Galmozzi, Michele Covella, Franco Veglio and Paolo Mulatero Department of Medicine and Experimental Oncology, Division of Internal Medicine and Hypertension, University of Torino, 10126 Torino, Italy Abstract: The last two decades have witnessed a growing number of experimental observations regarding the mineralocorticoid physiopathology, leading to a new understanding of the molecular basis of the aldosterone-induced target organ damage. As a matter of fact, although it has long been known that the combined administration of mineralocorticoids and salt leads to extensive vascular lesions in the target organs, the recognition that aldosterone is able to induce direct toxic effects on the various cell types that make up the cardiovascular organ has built up recently. Moreover, non-genomic effects have been attributed to aldosterone, i.e. effects which do not depend on the activation of the cellular transcription machinery, whose physiopathologic relevance is still being investigated. These advances in our understanding of aldosterone physiopathology have shed light on the biological reasons which are at the base of the impressive results obtained in clinical trials of aldosterone antagonism. Keywords: Aldosterone, hyperaldosteronism, endothelium. INTRODUCTION permeability by means of the generation of high electrochemical gradients. This action is performed by The hormone aldosterone was named after the aldehyde modulation of gene expression. group located on the carbon 18 of the steroidal skeleton which takes the place of the methyl group which Aldosterone synthase, the product of the CYP11B2 gene characterizes all the other steroidal compounds. -

Australian Product Information – Aldactone® (Spironolactone)
AUSTRALIAN PRODUCT INFORMATION – ALDACTONE® (SPIRONOLACTONE) 1. NAME OF THE MEDICINE Spironolactone 2. QUALITATIVE AND QUANTITATIVE COMPOSITION The 25 mg ALDACTONE tablets contain 25 mg spironolactone. The 100 mg ALDACTONE tablets contain 100 mg spironolactone. For the full list of excipients, see Section 6.1 List of excipients. 3. PHARMACEUTICAL FORM Film-coated tablets. 25 mg ALDACTONE tablets: 8.7 mm in diameter, round, biconvex, buff coloured, peppermint flavoured, film coated, stamped SEARLE over 39 on one side and unmarked on the other. 100 mg ALDACTONE tablets: 11.2 mm in diameter, round, biconvex, buff coloured, peppermint flavoured, film coated, stamped SEARLE over 134 on one side and unmarked on the other. 4. CLINICAL PARTICULARS 4.1 Therapeutic indications Essential Hypertension ALDACTONE, when used alone, is effective in lowering both systolic and diastolic blood pressure. ALDACTONE improves the hypotensive action of thiazide diuretics while at the same time reducing or preventing potassium loss due to the thiazide. ALDACTONE enhances the effectiveness of other antihypertensive agents such as beta blockers, vasodilators, etc. Version: pfpaldat10621 Supersedes: pfpaldat10220 Page 1 of 12 As adjunctive therapy in malignant hypertension. In diuretic-induced hypokalaemia when other measures are considered inappropriate or inadequate. Prophylaxis of hypokalaemia in patients taking digitalis when other measures are considered inadequate or inappropriate. Oedematous disorders, such as oedema and ascites of congestive cardiac failure, cirrhosis of the liver, nephrotic syndrome. Congestive Cardiac Failure ALDACTONE, when used alone, is effective in the management of oedema and sodium retention associated with congestive cardiac failure. ALDACTONE may be used in combination with a thiazide or other conventional diuretics for achieving diuresis in patients whose oedema is resistant to a thiazide or other conventional diuretics. -

(12) Patent Application Publication (10) Pub. No.: US 2002/0102215 A1 100 Ol
US 2002O102215A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2002/0102215 A1 Klaveness et al. (43) Pub. Date: Aug. 1, 2002 (54) DIAGNOSTIC/THERAPEUTICAGENTS (60) Provisional application No. 60/049.264, filed on Jun. 6, 1997. Provisional application No. 60/049,265, filed (75) Inventors: Jo Klaveness, Oslo (NO); Pal on Jun. 6, 1997. Provisional application No. 60/049, Rongved, Oslo (NO); Anders Hogset, 268, filed on Jun. 7, 1997. Oslo (NO); Helge Tolleshaug, Oslo (NO); Anne Naevestad, Oslo (NO); (30) Foreign Application Priority Data Halldis Hellebust, Oslo (NO); Lars Hoff, Oslo (NO); Alan Cuthbertson, Oct. 28, 1996 (GB)......................................... 9622.366.4 Oslo (NO); Dagfinn Lovhaug, Oslo Oct. 28, 1996 (GB). ... 96223672 (NO); Magne Solbakken, Oslo (NO) Oct. 28, 1996 (GB). 9622368.0 Jan. 15, 1997 (GB). ... 97OO699.3 Correspondence Address: Apr. 24, 1997 (GB). ... 9708265.5 BACON & THOMAS, PLLC Jun. 6, 1997 (GB). ... 9711842.6 4th Floor Jun. 6, 1997 (GB)......................................... 97.11846.7 625 Slaters Lane Alexandria, VA 22314-1176 (US) Publication Classification (73) Assignee: NYCOMED IMAGING AS (51) Int. Cl." .......................... A61K 49/00; A61K 48/00 (52) U.S. Cl. ............................................. 424/9.52; 514/44 (21) Appl. No.: 09/765,614 (22) Filed: Jan. 22, 2001 (57) ABSTRACT Related U.S. Application Data Targetable diagnostic and/or therapeutically active agents, (63) Continuation of application No. 08/960,054, filed on e.g. ultrasound contrast agents, having reporters comprising Oct. 29, 1997, now patented, which is a continuation gas-filled microbubbles stabilized by monolayers of film in-part of application No. 08/958,993, filed on Oct. -

Dosage and Administration
HGDS"'FG ·-···--··-· ..---- ·---·' .. e ·--··---····~·----- 133 Molesworth Street PO Box5013 Wellington 6140 New Zealand T +64 4 496 2000 1 February 2019 Response to your request for official information I refer to your request of 4 January 2019 to the Ministry of Health (the Ministry), under the Official Information Act 1982 (the Act), for. "I wish to request the following information regarding (the now lapsed) Aldactone, film coated tablets, spironolactone 25 mg and 100mg (TTS0-1764, 1764a): • Please provide a copy of the most recent datasheet (approved in a CMN or notified ina SACN). • Please provide a copy of the most recent primary and secondary labelling (approved in a CMN or notified in a SACN). • Please advise if the approved NZ labelling included an ARTG number." Information held by the Ministry relating to your request is itemised below, with copies of documents attached. Attachment Details and decision number - Data sheet for Aldactone 25 mg and Aldactone 100 mg. 1 Information released in full. Copy of the primary and secondary labelling for Aldactone 25 mg and 2 Aldactone 100 mg. Information released in full. In response to part three of your request, the Ministry confirms that the approved secondary labelling for Aldactone 25 mg and Aldactone 100 mg in New Zealand contained Australian Register of Therapeutic Goods (ARTG) numbers: AUST R 68953 (Aldactone 25 mg) and AUST R 68954 (Aldactone 100 mg). I trust this information fulfils your request. Please note this response (with your personal details removed) may be published on the Ministry of Health website. Yours si r;,c;:arely ~ r,,..~-~/___/ / /.. -

Federal Register / Vol. 60, No. 80 / Wednesday, April 26, 1995 / Notices DIX to the HTSUS—Continued
20558 Federal Register / Vol. 60, No. 80 / Wednesday, April 26, 1995 / Notices DEPARMENT OF THE TREASURY Services, U.S. Customs Service, 1301 TABLE 1.ÐPHARMACEUTICAL APPEN- Constitution Avenue NW, Washington, DIX TO THE HTSUSÐContinued Customs Service D.C. 20229 at (202) 927±1060. CAS No. Pharmaceutical [T.D. 95±33] Dated: April 14, 1995. 52±78±8 ..................... NORETHANDROLONE. A. W. Tennant, 52±86±8 ..................... HALOPERIDOL. Pharmaceutical Tables 1 and 3 of the Director, Office of Laboratories and Scientific 52±88±0 ..................... ATROPINE METHONITRATE. HTSUS 52±90±4 ..................... CYSTEINE. Services. 53±03±2 ..................... PREDNISONE. 53±06±5 ..................... CORTISONE. AGENCY: Customs Service, Department TABLE 1.ÐPHARMACEUTICAL 53±10±1 ..................... HYDROXYDIONE SODIUM SUCCI- of the Treasury. NATE. APPENDIX TO THE HTSUS 53±16±7 ..................... ESTRONE. ACTION: Listing of the products found in 53±18±9 ..................... BIETASERPINE. Table 1 and Table 3 of the CAS No. Pharmaceutical 53±19±0 ..................... MITOTANE. 53±31±6 ..................... MEDIBAZINE. Pharmaceutical Appendix to the N/A ............................. ACTAGARDIN. 53±33±8 ..................... PARAMETHASONE. Harmonized Tariff Schedule of the N/A ............................. ARDACIN. 53±34±9 ..................... FLUPREDNISOLONE. N/A ............................. BICIROMAB. 53±39±4 ..................... OXANDROLONE. United States of America in Chemical N/A ............................. CELUCLORAL. 53±43±0 -
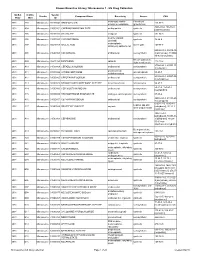
Known Bioactive Library: Microsource 1 - US Drug Collection
Known Bioactive Library: Microsource 1 - US Drug Collection ICCB-L ICCB-L Vendor Vendor Compound Name Bioactivity Source CAS Plate Well ID antifungal, inhibits Penicillium 2091 A03 Microsource 00200046 GRISEOFULVIN 126-07-8 mitosis in metaphase griseofulvum 3505-38-2, 486-16-8 2091 A04 Microsource 01500161 CARBINOXAMINE MALEATE antihistaminic synthetic [carbinoxamine] 2091 A05 Microsource 00200331 SALSALATE analgesic synthetic 552-94-3 muscle relaxant 2091 A06 Microsource 01500162 CARISOPRODOL synthetic 78-44-4 (skeletal) antineoplastic, 2091 A07 Microsource 00210369 GALLIC ACID insect galls 149-91-7 astringent, antibacterial 66592-87-8, 50370-12- 2091 A08 Microsource 01500163 CEFADROXIL antibacterial semisynthetic 2 [anhydrous], 119922- 89-9 [hemihydrate] Rheum palmatum, 2091 A09 Microsource 00211468 DANTHRON cathartic 117-10-2 Xyris semifuscata 27164-46-1, 25953-19- 2091 A10 Microsource 01500164 CEFAZOLIN SODIUM antibacterial semisynthetic 9 [cefazolin] glucocorticoid, 2091 A11 Microsource 00300024 HYDROCORTISONE adrenal glands 50-23-7 antiinflammatory 64485-93-4, 63527-52- 2091 A12 Microsource 01500165 CEFOTAXIME SODIUM antibacterial semisynthetic 6 [cefotaxime] 2091 A13 Microsource 00300029 DESOXYCORTICOSTERONE ACETATE mineralocorticoid adrenocortex 56-47-3 58-71-9, 153-61-7 2091 A14 Microsource 01500166 CEPHALOTHIN SODIUM antibacterial semisynthetic [cephalothin] 2091 A15 Microsource 00300034 TESTOSTERONE PROPIONATE androgen, antineoplastic semisynthetic 57-85-2 24356-60-3, 21593-23- 2091 A16 Microsource 01500167 CEPHAPIRIN SODIUM -

Stembook 2018.Pdf
The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 WHO/EMP/RHT/TSN/2018.1 © World Health Organization 2018 Some rights reserved. This work is available under the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 IGO licence (CC BY-NC-SA 3.0 IGO; https://creativecommons.org/licenses/by-nc-sa/3.0/igo). Under the terms of this licence, you may copy, redistribute and adapt the work for non-commercial purposes, provided the work is appropriately cited, as indicated below. In any use of this work, there should be no suggestion that WHO endorses any specific organization, products or services. The use of the WHO logo is not permitted. If you adapt the work, then you must license your work under the same or equivalent Creative Commons licence. If you create a translation of this work, you should add the following disclaimer along with the suggested citation: “This translation was not created by the World Health Organization (WHO). WHO is not responsible for the content or accuracy of this translation. The original English edition shall be the binding and authentic edition”. Any mediation relating to disputes arising under the licence shall be conducted in accordance with the mediation rules of the World Intellectual Property Organization. Suggested citation. The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances. Geneva: World Health Organization; 2018 (WHO/EMP/RHT/TSN/2018.1). Licence: CC BY-NC-SA 3.0 IGO. Cataloguing-in-Publication (CIP) data. -

Prestwick Collection
(-) -Levobunolol hydrochloride (-)-Adenosine 3',5'-cyclic monophosphate (-)-Cinchonidine (-)-Eseroline fumarate salt (-)-Isoproterenol hydrochloride (-)-MK 801 hydrogen maleate (-)-Quinpirole hydrochloride (+) -Levobunolol hydrochloride (+)-Isoproterenol (+)-bitartrate salt (+,-)-Octopamine hydrochloride (+,-)-Synephrine (±)-Nipecotic acid (1-[(4-Chlorophenyl)phenyl-methyl]-4-methylpiperazine) (cis-) Nanophine (d,l)-Tetrahydroberberine (R) -Naproxen sodium salt (R)-(+)-Atenolol (R)-Propranolol hydrochloride (S)-(-)-Atenolol (S)-(-)-Cycloserine (S)-propranolol hydrochloride 2-Aminobenzenesulfonamide 2-Chloropyrazine 3-Acetamidocoumarin 3-Acetylcoumarin 3-alpha-Hydroxy-5-beta-androstan-17-one 6-Furfurylaminopurine 6-Hydroxytropinone Acacetin Acebutolol hydrochloride Aceclofenac Acemetacin Acenocoumarol Acetaminophen Acetazolamide Acetohexamide Acetopromazine maleate salt Acetylsalicylsalicylic acid Aconitine Acyclovir Adamantamine fumarate Adenosine 5'-monophosphate monohydrate Adiphenine hydrochloride Adrenosterone Ajmalicine hydrochloride Ajmaline Albendazole Alclometasone dipropionate Alcuronium chloride Alexidine dihydrochloride Alfadolone acetate Alfaxalone Alfuzosin hydrochloride Allantoin alpha-Santonin Alprenolol hydrochloride Alprostadil Althiazide Altretamine Alverine citrate salt Ambroxol hydrochloride Amethopterin (R,S) Amidopyrine Amikacin hydrate Amiloride hydrochloride dihydrate Aminocaproic acid Aminohippuric acid Aminophylline Aminopurine, 6-benzyl Amiodarone hydrochloride Amiprilose hydrochloride Amitryptiline hydrochloride -

United States Patent (19) 11 Patent Number: 4,684,635 Orentreich Et Al
United States Patent (19) 11 Patent Number: 4,684,635 Orentreich et al. (45) Date of Patent: Aug. 4, 1987 54: COMPOSITIONS AND METHODS FOR N. Orentreich et al., "Biology of Scaly Hair Growth," INHIBITING THE ACTION OF ANDROGENS 9 Clinics in Plastic Surgery (1982), 197-205. R. Aron-Brunetiere, "Aspects of Endocrinological 75 Inventors: Norman Orentreich, 140 E. 72, New Treatment," Hair Research (1981), 312-317. York, N.Y. 10021; Jonathan R. J. R. Rentoul, "Management of the Hirsuite Woman," Matias, Richmond Hill, N.Y. 22 Int'l J. of Derm. (1983), 265-272. 73) Assignee: Norman Orentreich, New York, N.Y. P. G. Nielsen, "Treatment of Moderate Idiopathic Hir sutism,' 165 Dermatologica (1982), 636-639. 21) Appl. No.: 846,498 F. Neuman et al., "Central Actions of Antiandrogens,' Androgens and Antiandrogens (1977), 163-177. 22 Filed: Mar. 27, 1986 F. J. Ebling, "Antiandrogens and Dermatology,” An drogens and Antiandrogens (1977), 341-349. Related U.S. Application Data W. I. P. Mainwaring, “Modes of Action of Antiandro I63) Continuation of Ser. No. 609,152, May 11, 1984, aban gens,' Androgens and Antiandrogens (1977), 151-161. doned. J. P. Raynaud et al., "Present Trends in Antiandrogen 51) Int. Cl............................................... A61K 31/56 Research,” Androgens and Antiandrogens (1977), 52 U.S. Cl. .................................... 514/170; 514/171; 28-293. 514/175; 514/177; 514/178 A. L. Southren et al., "Effect of Progestagens,' Andro 58) Field of Search ............... 514/177, 178, 170, 171, gens and Antiandrogens (1977), 267-279. 514/175 Primary Examiner-Leonard Schenkman Attorney, Agent, or Firm-Panitch Schwarze Jacobs and (56) References Cited Nadel U.S. -

(12) Patent Application Publication (10) Pub. No.: US 2004/0058896 A1 Dietrich Et Al
US 200400.58896A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2004/0058896 A1 Dietrich et al. (43) Pub. Date: Mar. 25, 2004 (54) PHARMACEUTICAL PREPARATION (30) Foreign Application Priority Data COMPRISING AN ACTIVE DISPERSED ON A MATRIX Dec. 7, 2000 (EP)........................................ OO126847.3 (76) Inventors: Rango Dietrich, Konstanz (DE); Publication Classification Rudolf Linder, Kontanz (DE); Hartmut Ney, Konstanz (DE) (51) Int. Cl." ...................... A61K 31156; A61K 31/4439 (52) U.S. Cl. ........................... 514/171; 514/179; 514/338 Correspondence Address: (57) ABSTRACT NATH & ASSOCATES PLLC 1030 FIFTEENTH STREET, N.W. The present invention relates to the field of pharmaceutical SIXTH FLOOR technology and describes a novel advantageous preparation WASHINGTON, DC 20005 (US) for an active ingredient. The novel preparation is Suitable for 9 producing a large number of pharmaceutical dosage forms. (21) Appl. No.: 10/433,398 In the new preparation an active ingredient is present essentially uniformly dispersed in an excipient matrix com (22) PCT Filed: Dec. 6, 2001 posed of one or more excipients Selected from the group of fatty alcohol, triglyceride, partial glyceride and fatty acid (86) PCT No.: PCT/EPO1/14307 eSter. US 2004/0058896 A1 Mar. 25, 2004 PHARMACEUTICAL PREPARATION 0008 Further subject matters are evident from the claims. COMPRISING AN ACTIVE DISPERSED ON A MATRIX 0009. The preparations for the purpose of the invention preferably comprise numerous individual units in which at least one active ingredient particle, preferably a large num TECHNICAL FIELD ber of active ingredient particles, is present in an excipient 0001. The present invention relates to the field of phar matrix composed of the excipients of the invention (also maceutical technology and describes a novel advantageous referred to as active ingredient units hereinafter). -

Council of Europe Committee of Ministers (Partial
COUNCIL OF EUROPE COMMITTEE OF MINISTERS (PARTIAL AGREEMENT IN THE SOCIAL AND PUBLIC HEALTH FIELD) RESOLUTION AP (82) 2 ON THE CLASSIFICATION OF MEDICINES WHICH ARE OBTAINABLE ONLY ON MEDICAL PRESCRIPTION (Adopted by the Committee of Ministers on 2 June 1982 at the 348th meeting of the Ministers' Deputies and superseding Resolution AP (77) 1) AND APPENDIX containing the list of medicines adopted by the Public Health Committee (Partial Agreement) updated to 31 October 1982 RESOLUTION AP (82) 2 ON THE CLASSIFICATION OF MEDICINES WHICH ARE OBTAINABLE ONLY ON MEDICAL PRESCRIPTION 1 (Adopted by the Committee of Ministers on 2 June 1982 at the 348th meeting of the Ministers' Deputies) The Representatives on the Committee of Ministers of Belgium, France, the Federal Republic of Germany, Italy, Luxembourg, the Netherlands, the United Kingdom of Great Britain and Northern Ireland, these states being parties to the Partial Agreement in the social and public health field, and the Representatives of Austria, Denmark, Ireland and Switzerland, states which have participated in the public health activities carried out within the above-mentioned Partial Agreement since 1 October 1974, 2 April 1968, 23 September 1969 and 5 May 1964, respectively, Considering that, under the terms of its Statute, the aim of the Council of Europe is to achieve a greater unity between its Members for the purpose of safeguarding and realising the ideals and principles which are their common heritage and facilitating their economic and social progress; Having regard to the