Cumulant-Based Network Analysis of Genotype-Phenotype
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Clinical, Molecular, and Immune Analysis of Dabrafenib-Trametinib
Supplementary Online Content Chen G, McQuade JL, Panka DJ, et al. Clinical, molecular and immune analysis of dabrafenib-trametinib combination treatment for metastatic melanoma that progressed during BRAF inhibitor monotherapy: a phase 2 clinical trial. JAMA Oncology. Published online April 28, 2016. doi:10.1001/jamaoncol.2016.0509. eMethods. eReferences. eTable 1. Clinical efficacy eTable 2. Adverse events eTable 3. Correlation of baseline patient characteristics with treatment outcomes eTable 4. Patient responses and baseline IHC results eFigure 1. Kaplan-Meier analysis of overall survival eFigure 2. Correlation between IHC and RNAseq results eFigure 3. pPRAS40 expression and PFS eFigure 4. Baseline and treatment-induced changes in immune infiltrates eFigure 5. PD-L1 expression eTable 5. Nonsynonymous mutations detected by WES in baseline tumors This supplementary material has been provided by the authors to give readers additional information about their work. © 2016 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 09/30/2021 eMethods Whole exome sequencing Whole exome capture libraries for both tumor and normal samples were constructed using 100ng genomic DNA input and following the protocol as described by Fisher et al.,3 with the following adapter modification: Illumina paired end adapters were replaced with palindromic forked adapters with unique 8 base index sequences embedded within the adapter. In-solution hybrid selection was performed using the Illumina Rapid Capture Exome enrichment kit with 38Mb target territory (29Mb baited). The targeted region includes 98.3% of the intervals in the Refseq exome database. Dual-indexed libraries were pooled into groups of up to 96 samples prior to hybridization. -

A Study of the Differential Expression Profiles of Keshan Disease Lncrna/Mrna Genes Based on RNA-Seq
421 Original Article A study of the differential expression profiles of Keshan disease lncRNA/mRNA genes based on RNA-seq Guangyong Huang1, Jingwen Liu2, Yuehai Wang1, Youzhang Xiang3 1Department of Cardiology, Liaocheng People’s Hospital of Shandong University, Liaocheng, China; 2School of Nursing, Liaocheng Vocational & Technical College, Liaocheng, China; 3Shandong Institute for Endemic Disease Control, Jinan, China Contributions: (I) Conception and design: G Huang, Y Xiang; (II) Administrative support: G Huang, Y Wang; (III) Provision of study materials or patients: G Huang, J Liu, Y Xiang; (IV) Collection and assembly of data: G Huang, J Liu; (V) Data analysis and interpretation: J Liu, Y Wang; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. Correspondence to: Guangyong Huang, MD, PhD. Department of Cardiology, Liaocheng People’s Hospital of Shandong University, No. 67 of Dongchang Street, Liaocheng 252000, China. Email: [email protected]. Background: This study aims to analyze the differential expression profiles of lncRNA in Keshan disease (KSD) and to explore the molecular mechanism of the disease occurrence and development. Methods: RNA-seq technology was used to construct the lncRNA/mRNA expression library of a KSD group (n=10) and a control group (n=10), and then Cuffdiff software was used to obtain the gene lncRNA/ mRNA FPKM value as the expression profile of lncRNA/mRNA. The fold changes between the two sets of samples were calculated to obtain differential lncRNA/mRNA expression profiles, and a bioinformatics analysis of differentially expressed genes was performed. Results: A total of 89,905 lncRNAs and 20,315 mRNAs were detected. -

Positive Selection, Relaxation, and Acceleration in the Evolution of the Human and Chimp Genome
Positive Selection, Relaxation, and Acceleration in the Evolution of the Human and Chimp Genome Leonardo Arbiza1, Joaquı´n Dopazo2, Herna´n Dopazo1* 1 Pharmacogenomics and Comparative Genomics Unit, Centro de Investigacio´nPrı´ncipe Felipe (CIPF), Valencia, Spain, 2 Functional Genomics Unit, Bioinformatics Department, Centro de Investigacio´nPrı´ncipe Felipe (CIPF), Valencia, Spain For years evolutionary biologists have been interested in searching for the genetic bases underlying humanness. Recent efforts at a large or a complete genomic scale have been conducted to search for positively selected genes in human and in chimp. However, recently developed methods allowing for a more sensitive and controlled approach in the detection of positive selection can be employed. Here, using 13,198 genes, we have deduced the sets of genes involved in rate acceleration, positive selection, and relaxation of selective constraints in human, in chimp, and in their ancestral lineage since the divergence from murids. Significant deviations from the strict molecular clock were observed in 469 human and in 651 chimp genes. The more stringent branch-site test of positive selection detected 108 human and 577 chimp positively selected genes. An important proportion of the positively selected genes did not show a significant acceleration in rates, and similarly, many of the accelerated genes did not show significant signals of positive selection. Functional differentiation of genes under rate acceleration, positive selection, and relaxation was not statistically significant between human and chimp with the exception of terms related to G-protein coupled receptors and sensory perception. Both of these were over-represented under relaxation in human in relation to chimp. -

The Hypothalamus As a Hub for SARS-Cov-2 Brain Infection and Pathogenesis
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. The hypothalamus as a hub for SARS-CoV-2 brain infection and pathogenesis Sreekala Nampoothiri1,2#, Florent Sauve1,2#, Gaëtan Ternier1,2ƒ, Daniela Fernandois1,2 ƒ, Caio Coelho1,2, Monica ImBernon1,2, Eleonora Deligia1,2, Romain PerBet1, Vincent Florent1,2,3, Marc Baroncini1,2, Florence Pasquier1,4, François Trottein5, Claude-Alain Maurage1,2, Virginie Mattot1,2‡, Paolo GiacoBini1,2‡, S. Rasika1,2‡*, Vincent Prevot1,2‡* 1 Univ. Lille, Inserm, CHU Lille, Lille Neuroscience & Cognition, DistAlz, UMR-S 1172, Lille, France 2 LaBoratorY of Development and PlasticitY of the Neuroendocrine Brain, FHU 1000 daYs for health, EGID, School of Medicine, Lille, France 3 Nutrition, Arras General Hospital, Arras, France 4 Centre mémoire ressources et recherche, CHU Lille, LiCEND, Lille, France 5 Univ. Lille, CNRS, INSERM, CHU Lille, Institut Pasteur de Lille, U1019 - UMR 8204 - CIIL - Center for Infection and ImmunitY of Lille (CIIL), Lille, France. # and ƒ These authors contriButed equallY to this work. ‡ These authors directed this work *Correspondence to: [email protected] and [email protected] Short title: Covid-19: the hypothalamic hypothesis 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Diversity and Regulatory Impact of Copy Number Variation in the Primate Macaca Fascicularis
Gschwind et al. BMC Genomics (2017) 18:144 DOI 10.1186/s12864-017-3531-y RESEARCHARTICLE Open Access Diversity and regulatory impact of copy number variation in the primate Macaca fascicularis Andreas R. Gschwind1,2, Anjali Singh3, Ulrich Certa3, Alexandre Reymond1* and Tobias Heckel3* Abstract Background: Copy number variations (CNVs) are a significant source of genetic diversity and commonly found in mammalian genomes. We have generated a genome-wide CNV map for Cynomolgus monkeys (Macaca fascicularis). This crab-eating macaque is the closest animal model to humans that is used in biomedical research. Results: We show that Cynomolgus monkey CNVs are in general much smaller in size than gene loci and are specific to the population of origin. Genome-wide expression data from five vitally important organs demonstrates that CNVs in close proximity to transcription start sites associate strongly with expression changes. Among these eQTL genes we find an overrepresentation of genes involved in metabolism, receptor activity, and transcription. Conclusion: These results provide evidence that CNVs shape tissue transcriptomes in monkey populations, potentially offering an adaptive advantage. We suggest that this genetic diversity should be taken into account when using Cynomolgus macaques as models. Keywords: Cynomolgus monkey, CNV, Gene expression, eQTL, Olfactory receptors Background genes they can alter gene dosage, disrupt coding se- Copy number variations (CNVs) are genetic differences quences or modify the level and timing of gene expres- in the normal population displayed as microscopically sion for genes within the CNV [9, 10] and on its flanks invisible deletions or amplifications of stretches of gen- [4, 11–15]. -
Explorations in Olfactory Receptor Structure and Function by Jianghai
Explorations in Olfactory Receptor Structure and Function by Jianghai Ho Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Hiroaki Matsunami, Supervisor ___________________________ Jorg Grandl, Chair ___________________________ Marc Caron ___________________________ Sid Simon ___________________________ [Committee Member Name] Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2014 ABSTRACT Explorations in Olfactory Receptor Structure and Function by Jianghai Ho Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Hiroaki Matsunami, Supervisor ___________________________ Jorg Grandl, Chair ___________________________ Marc Caron ___________________________ Sid Simon ___________________________ [Committee Member Name] An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2014 Copyright by Jianghai Ho 2014 Abstract Olfaction is one of the most primitive of our senses, and the olfactory receptors that mediate this very important chemical sense comprise the largest family of genes in the mammalian genome. It is therefore surprising that we understand so little of how olfactory receptors work. In particular we have a poor idea of what chemicals are detected by most of the olfactory receptors in the genome, and for those receptors which we have paired with ligands, we know relatively little about how the structure of these ligands can either activate or inhibit the activation of these receptors. Furthermore the large repertoire of olfactory receptors, which belong to the G protein coupled receptor (GPCR) superfamily, can serve as a model to contribute to our broader understanding of GPCR-ligand binding, especially since GPCRs are important pharmaceutical targets. -
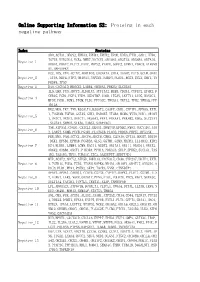
Online Supporting Information S2: Proteins in Each Negative Pathway
Online Supporting Information S2: Proteins in each negative pathway Index Proteins ADO,ACTA1,DEGS2,EPHA3,EPHB4,EPHX2,EPOR,EREG,FTH1,GAD1,HTR6, IGF1R,KIR2DL4,NCR3,NME7,NOTCH1,OR10S1,OR2T33,OR56B4,OR7A10, Negative_1 OR8G1,PDGFC,PLCZ1,PROC,PRPS2,PTAFR,SGPP2,STMN1,VDAC3,ATP6V0 A1,MAPKAPK2 DCC,IDS,VTN,ACTN2,AKR1B10,CACNA1A,CHIA,DAAM2,FUT5,GCLM,GNAZ Negative_2 ,ITPA,NEU4,NTF3,OR10A3,PAPSS1,PARD3,PLOD1,RGS3,SCLY,SHC1,TN FRSF4,TP53 Negative_3 DAO,CACNA1D,HMGCS2,LAMB4,OR56A3,PRKCQ,SLC25A5 IL5,LHB,PGD,ADCY3,ALDH1A3,ATP13A2,BUB3,CD244,CYFIP2,EPHX2,F CER1G,FGD1,FGF4,FZD9,HSD17B7,IL6R,ITGAV,LEFTY1,LIPG,MAN1C1, Negative_4 MPDZ,PGM1,PGM3,PIGM,PLD1,PPP3CC,TBXAS1,TKTL2,TPH2,YWHAQ,PPP 1R12A HK2,MOS,TKT,TNN,B3GALT4,B3GAT3,CASP7,CDH1,CYFIP1,EFNA5,EXTL 1,FCGR3B,FGF20,GSTA5,GUK1,HSD3B7,ITGB4,MCM6,MYH3,NOD1,OR10H Negative_5 1,OR1C1,OR1E1,OR4C11,OR56A3,PPA1,PRKAA1,PRKAB2,RDH5,SLC27A1 ,SLC2A4,SMPD2,STK36,THBS1,SERPINC1 TNR,ATP5A1,CNGB1,CX3CL1,DEGS1,DNMT3B,EFNB2,FMO2,GUCY1B3,JAG Negative_6 2,LARS2,NUMB,PCCB,PGAM1,PLA2G1B,PLOD2,PRDX6,PRPS1,RFXANK FER,MVD,PAH,ACTC1,ADCY4,ADCY8,CBR3,CLDN16,CPT1A,DDOST,DDX56 ,DKK1,EFNB1,EPHA8,FCGR3A,GLS2,GSTM1,GZMB,HADHA,IL13RA2,KIR2 Negative_7 DS4,KLRK1,LAMB4,LGMN,MAGI1,NUDT2,OR13A1,OR1I1,OR4D11,OR4X2, OR6K2,OR8B4,OXCT1,PIK3R4,PPM1A,PRKAG3,SELP,SPHK2,SUCLG1,TAS 1R2,TAS1R3,THY1,TUBA1C,ZIC2,AASDHPPT,SERPIND1 MTR,ACAT2,ADCY2,ATP5D,BMPR1A,CACNA1E,CD38,CYP2A7,DDIT4,EXTL Negative_8 1,FCER1G,FGD3,FZD5,ITGAM,MAPK8,NR4A1,OR10V1,OR4F17,OR52D1,O R8J3,PLD1,PPA1,PSEN2,SKP1,TACR3,VNN1,CTNNBIP1 APAF1,APOA1,CARD11,CCDC6,CSF3R,CYP4F2,DAPK1,FLOT1,GSTM1,IL2 -

Supplemental Table S18. Cellular Process Enrichment Analysis Output
Supplemental Table S18. -
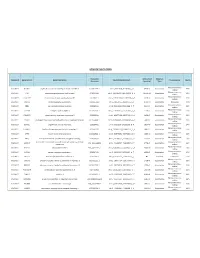
Somatic Mutations
SOMATIC MUTATIONS Transcript Amino Acid Mutation Sample ID Gene Symbol Gene Description Nucleotide (genomic) Consequence Mut % Accession (protein) Type Nonsynonymous PGDX11T ACSBG2 acyl-CoA synthetase bubblegum family member 2 CCDS12159.1 chr19_6141628_6141628_C_A 654A>D Substitution 46% coding Nonsynonymous PGDX11T ATR ataxia telangiectasia and Rad3 related CCDS3124.1 chr3_143721232_143721232_G_A 1451R>W Substitution 32% coding Nonsynonymous PGDX11T C1orf183 chromosome 1 open reading frame 183 CCDS841.1 chr1_112071336_112071336_C_T 224R>Q Substitution 22% coding PGDX11T CMYA5 cardiomyopathy associated 5 NM_153610 chr5_79063455_79063455_C_A 1037C>X Substitution Nonsense 34% Nonsynonymous PGDX11T CNR1 cannabinoid receptor 1 (brain) CCDS5015.1 chr6_88911646_88911646_C_T 23V>M Substitution 35% coding Nonsynonymous PGDX11T COL4A4 collagen; type IV; alpha 4 CCDS42828.1 chr2_227681807_227681807_G_A 227R>C Substitution 20% coding Nonsynonymous PGDX11T CYBASC3 cytochrome b; ascorbate dependent 3 CCDS8004.1 chr11_60877136_60877136_G_A 149R>C Substitution 34% coding Nonsynonymous PGDX11T DYRK3 dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 3 CCDS30999.1 chr1_204888091_204888091_G_A 309V>I Substitution 44% coding Nonsynonymous PGDX11T ELMO1 engulfment and cell motility 1 CCDS5449.1 chr7_37239295_37239295_G_A 160T>M Substitution 24% coding Nonsynonymous PGDX11T FAM83H family with sequence similarity 83; member H CCDS6410.2 chr8_144884473_144884473_C_A 90G>C Substitution 53% coding Nonsynonymous PGDX11T KPTN kaptin (actin binding protein) -

Supplementary Table S1. Genomic Context of Microdeletions Found in All Epilepsy Samples
Supplementary Table S1. Genomic context of microdeletions found in all epilepsy samples. Chr End Size Start Genes Probes N°Genes Overlaps SampleID HOTSPOT? Phenotype ACAP3,AGRN,ANKRD65,ATAD3A,ATAD3B,ATAD3C,AURKAIP1,B3GAL T6,C1orf159,C1orf233,CCNL2,CDK11B,CPSF3L,CPTP,DVL1,FAM132A ,HES4,ISG15,KLHL17,MIB2,MMP23B,MRPL20,MXRA8,NOC2L,PERM 1,PLEKHN1,PUSL1,RNF223,SAMD11,SCNN1D,SDF4,SLC35E2B,SSU72 ,TAS1R3,TMEM240,TMEM88B,TNFRSF18,TNFRSF4,TTLL10,UBE2J2,V WA1,LINC01342,LOC100130417,LOC102724312,LOC148413,MIR20 1 846808 4679951 3833144 1112 94 0A,MIR200B,MIR429,MIR6726,MIR6727,MIR6808,MMP23A,ACTRT IT-PR-2 AFE NO 2,C1orf86,CALML6,CFAP74,FAM213B,GABRD,GNB1,HES5,MMEL1, MORN1,NADK,PANK4,PEX10,PLCH2,PRDM16,PRKCZ,RER1,SKI,TME M52,TNFRSF14,TTC34,LINC00982,LOC100129534,LOC100996583,L OC115110,MIR4251,ARHGEF16,C1orf174,CCDC27,CEP104,DFFB,LR RC47,MEGF6,SMIM1,TP73,TPRG1L,WRAP73,LINC01134,MIR551A,T P73-AS1,LINC01346,LOC284661 1 4529544 5043734 514191 266 1 AJAP1 E472 RE NO 1 18361468 18853490 492023 419 3 IGSF21,KLHDC7A,LOC101927876 CTR-0001 Ctrl NO 1 50002235 50676365 674131 289 2 AGBL4,ELAVL4 CTR-0002 Ctrl NO 1 76631270 77036326 405057 294 1 ST6GALNAC3 CTR-0003 Ctrl NO 1 80073991 81847639 1773649 1198 1 LOC101927412 CTR-0004 Ctrl NO 1 81563249 82131272 568024 497 1 LOC101927434 CTR-0005 Ctrl NO 1 97005643 97712686 707044 389 3 DPYD,PTBP2,DPYD-AS1 EC-CAE428 GGE NO CDC14A,DBT,DPH5,EXTL2,GPR88,RTCA,SLC30A7,VCAM1,LINC01349 1 100661874 101502850 840977 488 12 CTR-0006 Ctrl NO ,LOC102606465,MIR553,RTCA-AS1 1 104452958 106299533 1846576 1037 2 LOC100129138,LOC101928476 -

( 12 ) United States Patent
US010167509B2 (12 ) United States Patent ( 10 ) Patent No. : US 10 , 167, 509 B2 Regan et al. ( 45 ) Date of Patent : Jan . 1 , 2019 (54 ) ANALYSIS OF NUCLEIC ACIDS ( 56 ) References Cited (71 ) Applicant: Bio - Rad Laboratories , Inc ., Hercules , U . S . PATENT DOCUMENTS CA (US ) 4 ,437 ,975 A 3 / 1984 Gillespie et al. 4 ,683 , 194 A * 7 / 1987 Saiki et al . .. 435 /6 . 12 (72 ) Inventors : John F . Regan , San Mateo , CA ( US ) ; 4 ,683 , 202 A 7 / 1987 Mullis Serge Saxonov , Oakland , CA (US ) ; 4 , 883 , 750 A 11/ 1989 Whiteley et al . 4 ,988 ,617 A 1 / 1991 Landegren et al . Michael Y . Lucero , San Francisco , CA 5 ,242 , 794 A 9 / 1993 Whiteley et al . ( US ) ; Benjamin J . Hindson , 5 ,409 , 818 A 4 / 1995 Davey et al. Livermore , CA (US ); Phillip 5 ,413 , 909 A 5 / 1995 Bassam et al . Belgrader , Severna Park , MD ( US) ; 5 , 426 , 180 A 6 / 1995 Kool 5 ,432 , 054 A 7 / 1995 Saunders et al. Simant Dube , Kirkland , WA (US ) ; 5 ,476 , 930 A 12 / 1995 Letsinger et al. Austin P . So , Pleasanton , CA (US ) ; 5 , 494 , 810 A 2 / 1996 Barany et al. Jeffrey C . Mellen , San Francisco , CA 5 ,554 ,517 A 9 / 1996 Davey et al. ( US ) ; Nicholas J . Heredia , Mountain 5 , 593 , 826 A 1 / 1997 Fung et al. House , CA (US ) ; Kevin D . Ness , 5 , 786 , 146 A 7 / 1998 Herman et al . 5 , 861 , 245 A 1 / 1999 Mcclelland et al . Pleasanton , CA (US ) ; Billy W . Colston , 5 ,866 ,337 A 2 / 1999 Schon Jr. -

Identifying Drug Sensitivity Subnetworks with NETPHIX
bioRxiv preprint doi: https://doi.org/10.1101/543876. this version posted February 8, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. It is made available under a CC-BY-NC-ND 4.0 International license. Identifying Drug Sensitivity Subnetworks with NETPHLIX Yoo-Ah Kim *† Rebecca Sarto Basso *†‡ Damian Wojtowicz † Dorit S. Hochbaum ‡ Fabio Vandin §¶ Teresa M. Prztycka †¶ Abstract Phenotypic heterogeneity in cancer is often caused by different patterns of genetic alterations. Under- standing such phenotype-genotype relationships is fundamental for the advance of personalized medicine. One of the important challenges in the area is to predict drug response on a personalized level. The pathway- centric view of cancer significantly advanced the understanding of genotype-phenotype relationships. How- ever, most of network identification methods in cancer focus on identifying subnetworks that include gen- eral cancer drivers or are associated with discrete features such as cancer subtypes, hence cannot be applied directly for the analysis of continuous features like drug response. On the other hand, existing genome wide association approaches do not fully utilize the complex proprieties of cancer mutational landscape. To ad- dress these challenges, we propose a computational method, named NETPHLIX (NETwork-to-PHenotpe mapping LeveragIng eXlusivity), which aims to identify mutated subnetworks that are associated with drug response (or any continuous cancer phenotype). Utilizing properties such as mutual exclusivity and interac- tions among genes, we formulate the problem as an integer linear program and solve it optimally to obtain a set of genes satisfying the constraints. NETPHLIX identified gene modules significantly associated with many drugs, including interesting response modules to MEK1/2 inhibitors in both directions (increased and decreased sensitivity to the drug) that the previous method, which does not utilize network informa- tion, failed to identify.