Identification and Characterization of a Novel Transcription
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

DULIP: a Dual Luminescence-Based Co-Immunoprecipitation Assay for Interactome Mapping in Mammalian Cells
Repository of the Max Delbrück Center for Molecular Medicine (MDC) in the Helmholtz Association http://edoc.mdc-berlin.de/14998 DULIP: A dual luminescence-based co-immunoprecipitation assay for interactome mapping in mammalian cells Trepte, P., Buntru, A., Klockmeier, K., Willmore, L., Arumughan, A., Secker, C., Zenkner, M., Brusendorf, L., Rau, K., Redel, A., Wanker, E.E. NOTICE: this is the author’s version of a work that was accepted for publication in the Journal of Molecular Biology. Changes resulting from the publishing process, such as peer review, editing, corrections, structural formatting, and other quality control mechanisms may not be reflected in this document. Changes may have been made to this work since it was submitted for publication. A definitive version was subsequently published in: Journal of Molecular Biology 2015 MMM DD ; 427(21): 3375-3388 doi: 10.1016/j.jmb.2015.08.003 Publisher: Elsevier © 2015, Elsevier. This work is licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/ or send a letter to Creative Commons, PO Box 1866, Mountain View, CA 94042, USA. DULIP: A DUAL LUMINESCENCE-BASED CO-IMMUNOPRECIPITATION ASSAY FOR INTERACTOME MAPPING IN MAMMALIAN CELLS Philipp Treptea, Alexander Buntrua#, Konrad Klockmeiera#, Lindsay Willmorea, Anup Arumughana, Christopher Seckera, Martina Zenknera, Lydia Brusendorfa, Kirstin Raua, Alexandra Redela and Erich E Wankera* a Neuroproteomics, Max Delbrueck Center for Molecular Medicine, Robert-Roessle- Straße 10, 13125 Berlin, Germany # Contributed equally * Corresponding author, E-mail address: [email protected], Telephone: +49- 30-9406-2157, Fax: +49-30-9406-2552 ABSTRACT Mapping of protein-protein interactions (PPIs) is critical for understanding protein function and complex biological processes. -

Supplementary Figure S4
18DCIS 18IDC Supplementary FigureS4 22DCIS 22IDC C D B A E (0.77) (0.78) 16DCIS 14DCIS 28DCIS 16IDC 28IDC (0.43) (0.49) 0 ADAMTS12 (p.E1469K) 14IDC ERBB2, LASP1,CDK12( CCNE1 ( NUTM2B SDHC,FCGR2B,PBX1,TPR( CD1D, B4GALT3, BCL9, FLG,NUP21OL,TPM3,TDRD10,RIT1,LMNA,PRCC,NTRK1 0 ADAMTS16 (p.E67K) (0.67) (0.89) (0.54) 0 ARHGEF38 (p.P179Hfs*29) 0 ATG9B (p.P823S) (0.68) (1.0) ARID5B, CCDC6 CCNE1, TSHZ3,CEP89 CREB3L2,TRIM24 BRAF, EGFR (7p11); 0 ABRACL (p.R35H) 0 CATSPER1 (p.P152H) 0 ADAMTS18 (p.Y799C) 19q12 0 CCDC88C (p.X1371_splice) (0) 0 ADRA1A (p.P327L) (10q22.3) 0 CCNF (p.D637N) −4 −2 −4 −2 0 AKAP4 (p.G454A) 0 CDYL (p.Y353Lfs*5) −4 −2 Log2 Ratio Log2 Ratio −4 −2 Log2 Ratio Log2 Ratio 0 2 4 0 2 4 0 ARID2 (p.R1068H) 0 COL27A1 (p.G646E) 0 2 4 0 2 4 2 EDRF1 (p.E521K) 0 ARPP21 (p.P791L) ) 0 DDX11 (p.E78K) 2 GPR101, p.A174V 0 ARPP21 (p.P791T) 0 DMGDH (p.W606C) 5 ANP32B, p.G237S 16IDC (Ploidy:2.01) 16DCIS (Ploidy:2.02) 14IDC (Ploidy:2.01) 14DCIS (Ploidy:2.9) -3 -2 -1 -3 -2 -1 -3 -2 -1 -3 -2 -1 -3 -2 -1 -3 -2 -1 Log Ratio Log Ratio Log Ratio Log Ratio 12DCIS 0 ASPM (p.S222T) Log Ratio Log Ratio 0 FMN2 (p.G941A) 20 1 2 3 2 0 1 2 3 2 ERBB3 (p.D297Y) 2 0 1 2 3 20 1 2 3 0 ATRX (p.L1276I) 20 1 2 3 2 0 1 2 3 0 GALNT18 (p.F92L) 2 MAPK4, p.H147Y 0 GALNTL6 (p.E236K) 5 C11orf1, p.Y53C (10q21.2); 0 ATRX (p.R1401W) PIK3CA, p.H1047R 28IDC (Ploidy:2.0) 28DCIS (Ploidy:2.0) 22IDC (Ploidy:3.7) 22DCIS (Ploidy:4.1) 18IDC (Ploidy:3.9) 18DCIS (Ploidy:2.3) 17q12 0 HCFC1 (p.S2025C) 2 LCMT1 (p.S34A) 0 ATXN7L2 (p.X453_splice) SPEN, p.P677Lfs*13 CBFB 1 2 3 4 5 6 7 8 9 10 11 -

Supplementary File 2A Revised
Supplementary file 2A. Differentially expressed genes in aldosteronomas compared to all other samples, ranked according to statistical significance. Missing values were not allowed in aldosteronomas, but to a maximum of five in the other samples. Acc UGCluster Name Symbol log Fold Change P - Value Adj. P-Value B R99527 Hs.8162 Hypothetical protein MGC39372 MGC39372 2,17 6,3E-09 5,1E-05 10,2 AA398335 Hs.10414 Kelch domain containing 8A KLHDC8A 2,26 1,2E-08 5,1E-05 9,56 AA441933 Hs.519075 Leiomodin 1 (smooth muscle) LMOD1 2,33 1,3E-08 5,1E-05 9,54 AA630120 Hs.78781 Vascular endothelial growth factor B VEGFB 1,24 1,1E-07 2,9E-04 7,59 R07846 Data not found 3,71 1,2E-07 2,9E-04 7,49 W92795 Hs.434386 Hypothetical protein LOC201229 LOC201229 1,55 2,0E-07 4,0E-04 7,03 AA454564 Hs.323396 Family with sequence similarity 54, member B FAM54B 1,25 3,0E-07 5,2E-04 6,65 AA775249 Hs.513633 G protein-coupled receptor 56 GPR56 -1,63 4,3E-07 6,4E-04 6,33 AA012822 Hs.713814 Oxysterol bining protein OSBP 1,35 5,3E-07 7,1E-04 6,14 R45592 Hs.655271 Regulating synaptic membrane exocytosis 2 RIMS2 2,51 5,9E-07 7,1E-04 6,04 AA282936 Hs.240 M-phase phosphoprotein 1 MPHOSPH -1,40 8,1E-07 8,9E-04 5,74 N34945 Hs.234898 Acetyl-Coenzyme A carboxylase beta ACACB 0,87 9,7E-07 9,8E-04 5,58 R07322 Hs.464137 Acyl-Coenzyme A oxidase 1, palmitoyl ACOX1 0,82 1,3E-06 1,2E-03 5,35 R77144 Hs.488835 Transmembrane protein 120A TMEM120A 1,55 1,7E-06 1,4E-03 5,07 H68542 Hs.420009 Transcribed locus 1,07 1,7E-06 1,4E-03 5,06 AA410184 Hs.696454 PBX/knotted 1 homeobox 2 PKNOX2 1,78 2,0E-06 -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Bioinformatics Analysis for the Identification of Differentially Expressed Genes and Related Signaling Pathways in H
Bioinformatics analysis for the identification of differentially expressed genes and related signaling pathways in H. pylori-CagA transfected gastric cancer cells Dingyu Chen*, Chao Li, Yan Zhao, Jianjiang Zhou, Qinrong Wang and Yuan Xie* Key Laboratory of Endemic and Ethnic Diseases , Ministry of Education, Guizhou Medical University, Guiyang, China * These authors contributed equally to this work. ABSTRACT Aim. Helicobacter pylori cytotoxin-associated protein A (CagA) is an important vir- ulence factor known to induce gastric cancer development. However, the cause and the underlying molecular events of CagA induction remain unclear. Here, we applied integrated bioinformatics to identify the key genes involved in the process of CagA- induced gastric epithelial cell inflammation and can ceration to comprehend the potential molecular mechanisms involved. Materials and Methods. AGS cells were transected with pcDNA3.1 and pcDNA3.1::CagA for 24 h. The transfected cells were subjected to transcriptome sequencing to obtain the expressed genes. Differentially expressed genes (DEG) with adjusted P value < 0.05, | logFC |> 2 were screened, and the R package was applied for gene ontology (GO) enrichment and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis. The differential gene protein–protein interaction (PPI) network was constructed using the STRING Cytoscape application, which conducted visual analysis to create the key function networks and identify the key genes. Next, the Submitted 20 August 2020 Kaplan–Meier plotter survival analysis tool was employed to analyze the survival of the Accepted 11 March 2021 key genes derived from the PPI network. Further analysis of the key gene expressions Published 15 April 2021 in gastric cancer and normal tissues were performed based on The Cancer Genome Corresponding author Atlas (TCGA) database and RT-qPCR verification. -
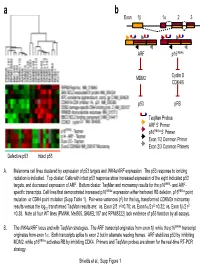
Taqman Probes ARF 5' Primer P16ink4a 5' Primer Exon 1/2
a b Exon 1β 1α 23 ARF p16INK4a MDM2 Cyclin D CDK4/6 p53 pRB TaqMan Probes ARF 5’ Primer p16INK4a 5’ Primer Exon 1/2 Common Primer Exon 2/3 Common Primers Defective p53 Intact p53 A. Melanoma cell lines clustered by expression of p53 targets and INK4a/ARF expression. The p53 response to ionizing radiation is indicated. Top cluster: Cells with intact p53 response show increased expression of the eight indicated p53 targets, and decreased expression of ARF. Bottom cluster: TaqMan and microarray results for the p16INK4- and ARF- specific transcripts. Cell lines that demonstrated increased p16INK4a expression either harbored RB deletion, p16INK4a point 2 mutation or CDK4 point mutation (Supp Table 1). Pair-wise variances (r ) for the log2 transformed CDKN2a microarray 2 2 2 results versus the log10 transformed TaqMan results are: vs. Exon 2/3 r =0.78; vs. Exon1α/2 r =0.32; vs. Exon 1β/2 r =0.38. Note all four WT lines (PMWK, Mel505, SKMEL187 and RPMI8322) lack evidence of p53 function by all assays. B. The INK4a/ARF locus and with TaqMan strategies. The ARF transcript originates from exon 1β while the p16INK4a transcript originates from exon 1α. Both transcripts splice to exon 2 but in alternate reading frames. ARF stabilizes p53 by inhibiting MDM2, while p16INK4a activates RB by inhibiting CDK4. Primers and TaqMan probes are shown for the real-time RT-PCR strategy. Shields et al., Supp Figure 1 SKMEL 28 U01 24h SKMEL WM2664 U01 48h WM2664 U01 24h 24 U01 48h SKMEL 24 U01 24h SKMEL 24 Untreated SKMEL 24 DMSO 48h SKMEL 24 DMSO 24h SKMEL WM2664 DMSO 24h WM2664 DMSO 48h WM2644 Untreated 28 Untreated SKMEL 28 DMSO 48h SKMEL 28 DMSO 24h SKMEL -3.00 -2.00 -1.00 0.00 1.00 2.00 3.00 relative to median expression Genes decreased by UO1 (863) HIF1A Hypoxia-inducible factor 1, alpha subunit NM_001530 RBBP8 Retinoblastoma binding protein 8 NM_002894 Homo sapiens, clone IMAGE:4337652, mRNA BC018676 EIF4EBP1 Eukaryotic translation initiation factor 4E binding protein EXOSC8 Exosome component 8 NM_181503 ENST00000321524 MCM7 MCM7 minichromosome maintenance deficient 7 (S. -
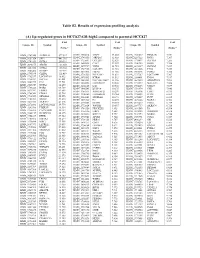
(A) Up-Regulated Genes in HCC827-GR-High2 Compared to Parental HCC827
Table S2. Results of expression profiling analysis (A) Up-regulated genes in HCC827-GR-high2 compared to parental HCC827 Fold Fold Fold Unique ID Symbol Unique ID Symbol Unique ID Symbol change* change* change* ILMN_1709348 ALDH1A1 577.587 ILMN_2310814 MAPT 13.003 ILMN_1741017 PIP4K2B 7.331 ILMN_1651354 SPP1 441.316 ILMN_1748650 MRPL45 12.988 ILMN_3237623 RNY1 7.297 ILMN_1701831 GSTA1 260.591 ILMN_1755897 UGT2B7 12.629 ILMN_1734897 SLC4A4 7.285 ILMN_1658835 CAV2 12.357 ILMN_1746359 RERG 7.280 ILMN_2094875 ABCB1 183.050 ILMN_1678939 VNN2 11.935 ILMN_1671337 SLC2A5 7.257 ILMN_3251540 GSTA2 145.982 ILMN_1729905 GAL3ST1 11.910 ILMN_1691606 LYG2 7.254 ILMN_2062468 IGFBP7 127.721 ILMN_1672536 FBLN1 11.716 ILMN_1785646 PMP22 7.246 ILMN_1795190 CLDN2 111.439 ILMN_1796339 PLEKHA2 11.631 ILMN_1737387 LOC728441 7.207 ILMN_1782937 LOC647169 98.612 ILMN_1676563 HTRA1 11.592 ILMN_1684401 FMO1 7.117 ILMN_1754247 SLC3A1 81.001 ILMN_3263423 LOC100129027 11.346 ILMN_1687035 ADAMTSL4 7.098 ILMN_1662795 CA2 79.581 ILMN_1694898 LOC653857 10.906 ILMN_2153572 MAGEA3 7.086 ILMN_2168747 GSTA2 66.250 ILMN_2404625 LAT 10.560 ILMN_1784283 USH1C 7.079 ILMN_1764228 DAB2 64.709 ILMN_1666546 DUSP14 10.375 ILMN_1731374 CPE 7.046 ILMN_1675797 EPDR1 63.605 ILMN_1764571 ARHGAP23 10.299 ILMN_1765446 EMP3 6.933 ILMN_1708341 PDZK1 59.714 ILMN_3200140 LOC645638 10.284 ILMN_1754002 IL1F8 6.863 ILMN_1713529 SEMA6A 52.575 ILMN_3244343 SNORA21 10.171 ILMN_1878007 FUT9 6.835 ILMN_1708391 NR1H4 43.218 ILMN_1671489 PC 10.075 ILMN_1699208 NAP1L1 6.763 ILMN_2412336 AKR1C2 42.826 ILMN_2404688 -

Whole Exome Sequencing Reveals NOTCH1 Mutations in Anaplastic Large Cell Lymphoma and Points to Notch Both As a Key Pathway and a Potential Therapeutic Target
Non-Hodgkin Lymphoma SUPPLEMENTARY APPENDIX Whole exome sequencing reveals NOTCH1 mutations in anaplastic large cell lymphoma and points to Notch both as a key pathway and a potential therapeutic target Hugo Larose, 1,2 Nina Prokoph, 1,2 Jamie D. Matthews, 1 Michaela Schlederer, 3 Sandra Högler, 4 Ali F. Alsulami, 5 Stephen P. Ducray, 1,2 Edem Nuglozeh, 6 Mohammad Feroze Fazaludeen, 7 Ahmed Elmouna, 6 Monica Ceccon, 2,8 Luca Mologni, 2,8 Carlo Gambacorti-Passerini, 2,8 Gerald Hoefler, 9 Cosimo Lobello, 2,10 Sarka Pospisilova, 2,10,11 Andrea Janikova, 2,11 Wilhelm Woessmann, 2,12 Christine Damm-Welk, 2,12 Martin Zimmermann, 13 Alina Fedorova, 14 Andrea Malone, 15 Owen Smith, 15 Mariusz Wasik, 2,16 Giorgio Inghirami, 17 Laurence Lamant, 18 Tom L. Blundell, 5 Wolfram Klapper, 19 Olaf Merkel, 2,3 G. A. Amos Burke, 20 Shahid Mian, 6 Ibraheem Ashankyty, 21 Lukas Kenner 2,3,22 and Suzanne D. Turner 1,2,10 1Department of Pathology, University of Cambridge, Cambridge, UK; 2European Research Initiative for ALK Related Malignancies (ERIA; www.ERIALCL.net ); 3Department of Pathology, Medical University of Vienna, Vienna, Austria; 4Unit of Laboratory Animal Pathology, Uni - versity of Veterinary Medicine Vienna, Vienna, Austria; 5Department of Biochemistry, University of Cambridge, Tennis Court Road, Cam - bridge, UK; 6Molecular Diagnostics and Personalised Therapeutics Unit, Colleges of Medicine and Applied Medical Sciences, University of Ha’il, Ha’il, Saudi Arabia; 7Neuroinflammation Research Group, Department of Neurobiology, A.I Virtanen Institute -

Overview Gene List Target Scan Vs DIANA Group a Group B Group A
Overview Gene list Target scan vs DIANA Group A Group B Group A hsa-miR-181a hsa-miR-323 hsa-miR-326 Target scan Diana microT Overlap Target scan Diana microT Overlap Target scan SEPT3 SEPT3 SEPT3 SEPT7 ADARB1 HPCAL4 ABHD2 ABL2 ABHD13 ACVR2A ADCYAP1R1 AKAP13 PDPK1 ACRBP ACAN ABI1 ADAMTS1 ALAD APOBR ACVRL1 ACCN2 ABLIM1 ADAMTSL1 ANKRD52 ATXN1 ADAM19 ACER3 ACSL1 AKAP7 ARID2 C18orf23,RNF165 ADAM33 ACVR2A ACTN2 ANKRD43 ARL3 C20orf29 ADAMTS2 ADAMTS1 ACVR2A AP1S3 ARRB1 CACNG4 AHCYL2 ADAMTS18 ACVR2B ARID2 BBC3 CCNJL ALOX15B ADAMTS5 ADAM11 ATP11A BTG1 CYP2E1 ANK1 ADAMTSL1 ADAM22 ATXN1 C18orf62 GNB1L ANKS6 ADARB1 ADAMTS1 B4GALT1 C1orf21 GPR61 APBA1 AFAP1 ADAMTS6 BAG4 CADM4 GTSE1 ARCN1 AFTPH ADAMTSL1 BAI3 CALML4 HPCAL4 ARHGEF37 AK3 ADCY9 BNC2 CAPN6 KIAA0152 ARID3B AKAP7 ADRBK1 BRD1 CBFA2T2 KIF1A ARL8A ANAPC16 AFF2 BRWD1 CEBPA MACF1 ATP2B2 ANK1 AHCTF1,AHCTF1PBTBD3 CHD1 MYO1D ATP6V1G2 ANKRD12 AKAP2,PALM2 C13orf23 CIT PCNT AUP1 ANKRD33B AKAP6 C14orf43 CLASP2 PDPK1 BCL2L2 ANKRD43 AKAP7 CAPRIN1 CLCN5 PLEKHG4B BHLHE40 ANKRD44 AKAP9 CARM1 CLIP3 PPARA BTBD3 ANKRD52 AKT3 CBX4 COL5A2 PRB1,PRB2,PRB4 BTRC AP1S3 ALG9 CCDC117 CTNS PTPRT C10orf26 APBA1 ANKRD13C CCNJ DCTN4 PYCR1 C14orf1 APLP2 ANKRD20B CDH13 DCUN1D4 RAPGEF1 C16orf45 APOO ANKRD43 CDON DDB1 SRCAP C16orf54 ARID2 ANKRD50 CDYL DDX39B TMEM63C C1orf106 ARL3 AP1G1 CEP350 DIP2C C1orf27 ARRDC3 AP1S3 CHD7 DNAJB3 C22orf29 ATF7 API5 CHIC1 EEPD1 C9orf3 ATG2B ARFGEF2 CLIP1 EIF2C1 CACNA1E ATG7 ARHGAP12 CNOT6L ELFN2 CAPN12 ATP11A ARHGAP26 CNR1 ELK1 CASKIN1 ATP2B3 ARHGAP29 CNTN4 FAM172A CBFA2T3 ATP8B2 ARHGEF3 CNTNAP2 -

Affymetrix Probeset ID Gene Symbol Gene Description
Affymetrix_ Gene_Symbol Gene_Description ProbeSet_ID 7896952 ATAD3A ATPase family, AAA domain containing 3A 7897068 SKI v-ski sarcoma viral oncogene homolog (avian) 7897132 PRDM16 PR domain containing 16 7897280 HES3 hairy and enhancer of split 3 (Drosophila) 7897737 C1orf187 chromosome 1 open reading frame 187 7898537 PAX7 paired box 7 7898693 ALPL alkaline phosphatase, liver/bone/kidney 7898739 CDC42 "cell division cycle 42 (GTP binding protein, 25kDa) " 7898799 C1QC "complement component 1, q subcomponent, C chain " 7898988 CLIC4 chloride intracellular channel 4 7899167 LIN28A lin-28 homolog A (C. elegans) 7899187 HMGN2 high-mobility group nucleosomal binding domain 2 7899265 SFN stratifin 7899562 PTPRU "protein tyrosine phosphatase, receptor type, U " 7899753 LCK lymphocyte-specific protein tyrosine kinase 7899774 HDAC1 histone deacetylase 1 7899790 TSSK3 testis-specific serine kinase 3 7900146 ZC3H12A zinc finger CCCH-type containing 12A 7900340 BMP8A bone morphogenetic protein 8a 7900699 CDC20 cell division cycle 20 homolog (S. cerevisiae) 7900792 PTPRF protein tyrosine phosphatase, receptor type, F 7901073 UROD uroporphyrinogen decarboxylase 7901123 NASP nuclear autoantigenic sperm protein (histone-binding) 7901140 MAST2 microtubule associated serine/threonine kinase 2 7901363 CDKN2C "cyclin-dependent kinase inhibitor 2C (p18, inhibits CDK4) " 7901557 DMRTB1 "DMRT-like family B with proline-rich C-terminal, 1 " 7901696 PCSK9 proprotein convertase subtilisin/kexin type 9 7901913 FOXD3 forkhead box D3 7902227 GADD45A growth arrest -

(12) Patent Application Publication (10) Pub. No.: US 2009/0269772 A1 Califano Et Al
US 20090269772A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2009/0269772 A1 Califano et al. (43) Pub. Date: Oct. 29, 2009 (54) SYSTEMS AND METHODS FOR Publication Classification IDENTIFYING COMBINATIONS OF (51) Int. Cl. COMPOUNDS OF THERAPEUTIC INTEREST CI2O I/68 (2006.01) CI2O 1/02 (2006.01) (76) Inventors: Andrea Califano, New York, NY G06N 5/02 (2006.01) (US); Riccardo Dalla-Favera, New (52) U.S. Cl. ........... 435/6: 435/29: 706/54; 707/E17.014 York, NY (US); Owen A. (57) ABSTRACT O'Connor, New York, NY (US) Systems, methods, and apparatus for searching for a combi nation of compounds of therapeutic interest are provided. Correspondence Address: Cell-based assays are performed, each cell-based assay JONES DAY exposing a different sample of cells to a different compound 222 EAST 41ST ST in a plurality of compounds. From the cell-based assays, a NEW YORK, NY 10017 (US) Subset of the tested compounds is selected. For each respec tive compound in the Subset, a molecular abundance profile from cells exposed to the respective compound is measured. (21) Appl. No.: 12/432,579 Targets of transcription factors and post-translational modu lators of transcription factor activity are inferred from the (22) Filed: Apr. 29, 2009 molecular abundance profile data using information theoretic measures. This data is used to construct an interaction net Related U.S. Application Data work. Variances in edges in the interaction network are used to determine the drug activity profile of compounds in the (60) Provisional application No. 61/048.875, filed on Apr.