Interaction Between Herpesviruses and Genetic Variation in Schizophrenia Pathogenesis: a Candidate Gene Approach
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Inflammation-Dependent IL18 Signaling Restricts Hepatocellular Carcinoma Growth by Enhancing the Accumulation and Activity of Tumor-Infiltrating Lymphocytes
Published OnlineFirst February 18, 2016; DOI: 10.1158/0008-5472.CAN-15-1548 Cancer Tumor and Stem Cell Biology Research Inflammation-Dependent IL18 Signaling Restricts Hepatocellular Carcinoma Growth by Enhancing the Accumulation and Activity of Tumor- Infiltrating Lymphocytes Geoffrey J. Markowitz1, Pengyuan Yang1,2,3, Jing Fu3, Gregory A. Michelotti4, Rui Chen1, Jianhua Sui5, Bin Yang2, Wen-Hao Qin3, Zheng Zhang6, Fu-Sheng Wang6, Anna Mae Diehl4, Qi-Jing Li7, Hongyang Wang3, and Xiao-Fan Wang1 Abstract Chronic inflammation in liver tissue is an underlying cause of IL18R1 deletion increased tumor burden. Mechanistically, we hepatocellular carcinoma. High levels of inflammatory cytokine foundthatIL18exertedinflammation-dependent tumor-sup- IL18 in the circulation of patients with hepatocellular carcinoma pressive effects largely by promoting the differentiation, activ- correlates with poor prognosis. However, conflicting results have ity, and survival of tumor-infiltrating T cells. Finally, differences been reported for IL18 in hepatocellular carcinoma development in the expression of IL18 in tumor tissue versus nontumor and progression. In this study, we used tissue specimens from tissueweremorepredictiveofpatientoutcomethanoverall hepatocellular carcinoma patients and clinically relevant mouse tissue expression. Taken together, our findings resolve a long- models of hepatocellular carcinoma to evaluate IL18 expression standing contradiction regarding a tumor-suppressive role for and function. In a mouse model of liver fibrosis that recapitulates IL18 in established hepatocellular carcinoma and provide a a tumor-promoting microenvironment, global deletion of the mechanistic explanation for the complex relationship between IL18 receptor IL18R1 enhanced tumor growth and burden. Sim- its expression pattern and hepatocellular carcinoma prognosis. ilarly, in a carcinogen-induced model of liver tumorigenesis, Cancer Res; 76(8); 1–12. -
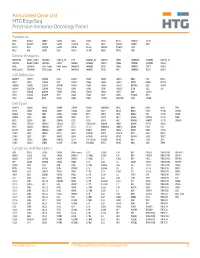
Annotated Gene List HTG Edgeseq Precision Immuno-Oncology Panel
Annotated Gene List HTG EdgeSeq Precision Immuno-Oncology Panel Apoptosis APAF1 BCL2L1 CARD11 CASP4 CD5L FADD KSR2 OPTN SAMD12 TCF19 BAX BCL2L11 CASP1 CASP5 CORO1A FAS LRG1 PLA2G6 SAMD9 XAF1 BCL10 BCL6 CASP10 CASP8 DAPK2 FASLG MECOM PYCARD SPOP BCL2 BID CASP3 CAV1 DAPL1 GLIPR1 MELK RIPK2 TBK1 Cancer Antigens ANKRD30A BAGE2_BAGE3 CEACAM6 CTAG1A_1B LIPE MAGEA3_A6 MAGEC2 PAGE3 SPANXACD SPANXN4 XAGE1B_1E ARMCX6 BAGE4_BAGE5 CEACAM8 CTAG2 MAGEA1 MAGEA4 MTFR2 PAGE4 SPANXB1 SPANXN5 XAGE2 BAGE CEACAM1 CT45_family GAGE_family MAGEA10 MAGEB2 PAGE1 PAGE5 SPANXN1 SYCP1 XAGE3 BAGE_family CEACAM5 CT47_family HPN MAGEA12 MAGEC1 PAGE2 PBK SPANXN3 TEX14 XAGE5 Cell Adhesion ADAM17 CDH15 CLEC5A DSG3 ICAM2 ITGA5 ITGB2 LAMC3 MBL2 PVR UPK2 ADD2 CDH5 CLEC6A DST ICAM3 ITGA6 ITGB3 LAMP1 MTDH RRAS2 UPK3A ADGRE5 CLDN3 CLEC7A EPCAM ICAM4 ITGAE ITGB4 LGALS1 NECTIN2 SELE VCAM1 ALCAM CLEC12A CLEC9A FBLN1 ITGA1 ITGAL ITGB7 LGALS3 OCLN SELL ZYX CD63 CLEC2B DIAPH3 FXYD5 ITGA2 ITGAM ITLN2 LYVE1 OLR1 SELPLG CD99 CLEC4A DLGAP5 IBSP ITGA3 ITGAX JAML M6PR PECAM1 THY1 CDH1 CLEC4C DSC3 ICAM1 ITGA4 ITGB1 L1CAM MADCAM1 PKP1 UNC5D Cell Cycle ANAPC1 CCND3 CDCA5 CENPH CNNM1 ESCO2 HORMAD2 KIF2C MELK ORC6 SKA3 TPX2 ASPM CCNE1 CDCA8 CENPI CNTLN ESPL1 IKZF1 KIF4A MND1 PATZ1 SP100 TRIP13 AURKA CCNE2 CDK1 CENPL CNTLN ETS1 IKZF2 KIF5C MYBL2 PIF1 SP110 TROAP AURKB CCNF CDK4 CENPU DBF4 ETS2 IKZF3 KIFC1 NCAPG PIMREG SPC24 TUBB BEX1 CDC20 CDK6 CENPW E2F2 EZH2 IKZF4 KNL1 NCAPG2 PKMYT1 SPC25 ZWILCH BEX2 CDC25A CDKN1A CEP250 E2F7 GADD45GIP1 KDM5B LMNA NCAPH POC1A SPDL1 BUB1 CDC25C CDKN1B CEP55 ECT2 -

Development of Autoimmune Hair Loss Disease Alopecia Areata Is Associated with Cardiac Dysfunction in C3H/ Hej Mice
Development of Autoimmune Hair Loss Disease Alopecia Areata Is Associated with Cardiac Dysfunction in C3H/ HeJ Mice Eddy Wang1, Katy Chong2, Mei Yu1, Noushin Akhoundsadegh1, David J. Granville3, Jerry Shapiro4, Kevin J. McElwee1* 1 Department of Dermatology and Skin Science, University of British Columbia, Vancouver, BC, Canada, 2 University of British Columbia, Vancouver, BC, Canada, 3 Department of Pathology and Laboratory Medicine, James Hogg Research Centre, Institute for Heart and Lung Health, University of British Columbia, Vancouver, BC, Canada, 4 Department of Dermatology and Skin Science, Vancouver General Hospital, Vancouver, BC, Canada Abstract Alopecia areata (AA) is a chronic autoimmune hair loss disease that affects several million men, women and children worldwide. Previous studies have suggested a link between autoimmunity, stress hormones, and increased cardiovascular disease risk. In the current study, histology, immunohistology, quantitative PCR (qPCR) and ELISAs were used to assess heart health in the C3H/HeJ mouse model for AA and heart tissue response to adrenocorticotropic hormone (ACTH) exposure. Mice with AA exhibited both atrial and ventricular hypertrophy, and increased collagen deposition compared to normal- haired littermates. QPCR revealed significant increases in Il18 (4.6-fold), IL18 receptor-1 (Il18r1; 2.8-fold) and IL18 binding protein (Il18bp; 5.2-fold) in AA hearts. Time course studies revealed a trend towards decreased Il18 in acute AA compared to controls while Il18r1, Il18bp and Casp1 showed similar trends to those of chronic AA affected mice. Immunohistochemistry showed localization of IL18 in chronic AA mouse atria. ELISA indicated cardiac troponin-I (cTnI) was elevated in the serum and significantly increased in AA heart tissue. -

Interleukin-18 in Health and Disease
International Journal of Molecular Sciences Review Interleukin-18 in Health and Disease Koubun Yasuda 1 , Kenji Nakanishi 1,* and Hiroko Tsutsui 2 1 Department of Immunology, Hyogo College of Medicine, 1-1 Mukogawa-cho, Nishinomiya, Hyogo 663-8501, Japan; [email protected] 2 Department of Surgery, Hyogo College of Medicine, 1-1 Mukogawa-cho, Nishinomiya, Hyogo 663-8501, Japan; [email protected] * Correspondence: [email protected]; Tel.: +81-798-45-6573 Received: 21 December 2018; Accepted: 29 January 2019; Published: 2 February 2019 Abstract: Interleukin (IL)-18 was originally discovered as a factor that enhanced IFN-γ production from anti-CD3-stimulated Th1 cells, especially in the presence of IL-12. Upon stimulation with Ag plus IL-12, naïve T cells develop into IL-18 receptor (IL-18R) expressing Th1 cells, which increase IFN-γ production in response to IL-18 stimulation. Therefore, IL-12 is a commitment factor that induces the development of Th1 cells. In contrast, IL-18 is a proinflammatory cytokine that facilitates type 1 responses. However, IL-18 without IL-12 but with IL-2, stimulates NK cells, CD4+ NKT cells, and established Th1 cells, to produce IL-3, IL-9, and IL-13. Furthermore, together with IL-3, IL-18 stimulates mast cells and basophils to produce IL-4, IL-13, and chemical mediators such as histamine. Therefore, IL-18 is a cytokine that stimulates various cell types and has pleiotropic functions. IL-18 is a member of the IL-1 family of cytokines. IL-18 demonstrates a unique function by binding to a specific receptor expressed on various types of cells. -

Fibrosis Accumulation in Idiopathic Pulmonary Apoptosis
Increased Cell Surface Fas Expression Is Necessary and Sufficient To Sensitize Lung Fibroblasts to Fas Ligation-Induced Apoptosis: Implications for Fibroblast This information is current as Accumulation in Idiopathic Pulmonary of October 1, 2021. Fibrosis Murry W. Wynes, Benjamin L. Edelman, Amanda G. Kostyk, Michael G. Edwards, Christopher Coldren, Steve D. Groshong, Gregory P. Cosgrove, Elizabeth F. Redente, Alison Bamberg, Kevin K. Brown, Nichole Reisdorph, Downloaded from Rebecca C. Keith, Stephen K. Frankel and David W. H. Riches J Immunol 2011; 187:527-537; Prepublished online 1 June 2011; doi: 10.4049/jimmunol.1100447 http://www.jimmunol.org/ http://www.jimmunol.org/content/187/1/527 Supplementary http://www.jimmunol.org/content/suppl/2011/06/01/jimmunol.110044 Material 7.DC1 References This article cites 62 articles, 18 of which you can access for free at: by guest on October 1, 2021 http://www.jimmunol.org/content/187/1/527.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2011 by The American Association of Immunologists, Inc. -

IL1R9 Is Evolutionarily Related to IL18BP and May Function As an IL-18 Receptor Chris S
IL1R9 Is Evolutionarily Related to IL18BP and May Function as an IL-18 Receptor Chris S. Booker and David R. Grattan This information is current as J Immunol published online 23 November 2016 of September 28, 2021. http://www.jimmunol.org/content/early/2016/11/23/jimmun ol.1500648 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2016/11/23/jimmunol.150064 Material 8.DCSupplemental Why The JI? Submit online. http://www.jimmunol.org/ • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average by guest on September 28, 2021 Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Published November 23, 2016, doi:10.4049/jimmunol.1500648 The Journal of Immunology IL1R9 Is Evolutionarily Related to IL18BP and May Function as an IL-18 Receptor Chris S. Booker and David R. Grattan The IL-1 families of ligands and receptors exhibit similarity of coding sequences, protein structures, and chromosomal positions, suggesting that they have arisen via duplication of ancestral genes. -

Production of IL-18 Binding Protein by Radiosensitive and Radioresistant
Production of IL-18 Binding Protein by Radiosensitive and Radioresistant Cells in CpG-Induced Macrophage Activation Syndrome This information is current as of October 4, 2021. Mathilde Harel, Charlotte Girard-Guyonvarc'h, Emiliana Rodriguez, Gaby Palmer and Cem Gabay J Immunol published online 10 July 2020 http://www.jimmunol.org/content/early/2020/07/09/jimmun ol.2000168 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2020/07/10/jimmunol.200016 Material 8.DCSupplemental http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on October 4, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2020 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Published July 10, 2020, doi:10.4049/jimmunol.2000168 The Journal of Immunology Production of IL-18 Binding Protein by Radiosensitive and Radioresistant Cells in CpG-Induced Macrophage Activation Syndrome Mathilde Harel,*,† Charlotte Girard-Guyonvarc’h,† Emiliana Rodriguez,*,† Gaby Palmer,*,†,1 and Cem Gabay*,†,1 IL-18 binding protein (IL-18BP) acts as a naturally occurring IL-18 decoy receptor. -

Clinical and Prognostic Relevance of CXCL12 Expression in Acute Myeloid Leukemia
Clinical and prognostic relevance of CXCL12 expression in acute myeloid leukemia Shi-sen Wang1,*, Zi-jun Xu2,3,*, Ye Jin1,3, Ji-chun Ma2,3, Pei-hui Xia2,3, Xiangmei Wen2,3, Zhen-wei Mao2, Jiang Lin2,3 and Jun Qian1,3 1 Department of Hematology, Affiliated People’s Hospital of Jiangsu University, Zhenjiang, Jiangsu, China 2 Laboratory Center, Affiliated People’s Hospital of Jiangsu University, Zhenjiang, Jiangsu, China 3 Zhenjiang Clinical Research Center of Hematology, Zhenjiang, Jiangsu, China * These authors contributed equally to this work. ABSTRACT Background: Accumulating studies have been made to understand the association between CXC chemokine ligand-12 (CXCL12)/CXC chemokine receptor 4 (CXCR4) and acute myeloid leukemia (AML). However, large-scale data analysis of potential relationship between CXCL12 and AML remains insufficient. Methods: We collected abundant CXCL12 expression data and AML samples from several publicly available datasets. The CIBERSORT algorithm was used to quantify immune cell fractions and the online website of STRING was utilized for gene ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis. The statistical analysis and graphical work were mainly performed via the R software. Results: CXCL12 expression was extremely down-regulated in AML. Clinically, low CXCL12 expression was correlated with higher white blood cells (WBCs) (P < 0.0001), more blasts in bone marrow (BM) (P < 0.001) and peripheral blood (PB) (P < 0.0001), FLT3-internal tandem duplications (FLT3-ITD) (P = 0.010) and NPM1 mutations (P = 0.015). More importantly, reduced CXCL12 expression Submitted 31 March 2021 Accepted 29 June 2021 predicted worse overall survival (OS) and event-free survival (EFS) in all AML, non- Published 20 July 2021 M3-AML, and cytogenetically normal (CN)-AML patients in three independent fi Corresponding authors cohorts. -

Supplementary Tables
SUPPLEMENTARY TABLES Supplementary Table 1. The clinicopathological features of the prognosis cohort associated with Foxp3 expression. (n = 72). Variables All patients Foxp3-positive Foxp3-negtive Age [years; n (%)] <60 62(86.1) 26(81.2) 36(94.7) ≥60 10(13.9) 8(18.8) 2(5.3) Gender [n (%)] Male 47(65.3) 24(70.6) 23(60.5) Female 25(45.7) 10(29.4) 15(39.5) WHO grade [n (%)] WHO III 49(68.1) 19(55.9) 30(78.9) WHO IV 23(31.9) 15(44.1) 8(21.1) Over survival 1179.1±1759.1 845.0±1386.8 1478.0±2007.0 (days, mean ±SD) Supplementary Table 2. The 29 immune signatures represented by 29 different gene sets. Immune signatures Genes aDCs CD83, LAMP3,CCL1 APC co inhibition C10orf54, CD274, LGALS9, PDCD1LG2, PVRL3, APC co stimulation CD40, CD58, CD70, ICOSLG, SLAMF1, TNFSF14, TNFSF15, TNFSF18, TNFSF4, TNFSF8, TNFSF9 B cells BACH2, BANK1, BLK, BTLA, CD79A, CD79B, FCRL1, FCRL3, HVCN1, RALGPS2 CCR CCL16, TPO, TGFBR2, CXCL2, CCL14, TGFBR3, IL11RA, CCL11, IL4I1, IL33, CXCL12, CXCL10, BMPER, BMP8A, CXCL11, IL21R, IL17B, TNFRSF9, ILF2, CX3CR1, CCR8, TNFSF12, CSF3, TNFSF4, BMP3, CX3CL1, BMP5, CXCR2, TNFRSF10D, BMP2, CXCL14, CCL28, CXCL3, BMP6, CCL21, CXCL9, CCL23, IL6, TNFRSF18, IL17RD, IL17D, IL27, CCL7, IL1R1, CXCR4, CXCR2P1, TGFB1I1, IFNGR1, IL9R, IL1RAPL1, IL11, CSF1, IL20RA, IL25, TNFRSF4, IL18, ILF3, CCL20, TNFRSF12A, IL6ST, CXCL13, IL12B, TNFRSF8, IL6R, BMPR2, IFNE, IL1RAPL2, IL3RA, BMP4, CCL24, TNFSF13B, CCR4, IL2RA, IL32, TNFRSF10C, IL22RA1, BMPR1A, CXCR5, CXCR3, IFNA8, IL17REL, IFNB1, IFNAR1, TNFRSF1B, CCL17, IFNL1, IL16, IL1RL1, ILK, CCL25, ILDR2, CXCR1, -

Autocrine IFN Signaling Inducing Profibrotic Fibroblast Responses By
Downloaded from http://www.jimmunol.org/ by guest on September 23, 2021 Inducing is online at: average * The Journal of Immunology , 11 of which you can access for free at: 2013; 191:2956-2966; Prepublished online 16 from submission to initial decision 4 weeks from acceptance to publication August 2013; doi: 10.4049/jimmunol.1300376 http://www.jimmunol.org/content/191/6/2956 A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Autocrine IFN Signaling Feng Fang, Kohtaro Ooka, Xiaoyong Sun, Ruchi Shah, Swati Bhattacharyya, Jun Wei and John Varga J Immunol cites 49 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130037 6.DC1 This article http://www.jimmunol.org/content/191/6/2956.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 23, 2021. The Journal of Immunology A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Inducing Autocrine IFN Signaling Feng Fang,* Kohtaro Ooka,* Xiaoyong Sun,† Ruchi Shah,* Swati Bhattacharyya,* Jun Wei,* and John Varga* Activation of TLR3 by exogenous microbial ligands or endogenous injury-associated ligands leads to production of type I IFN. -

1 the IL-18 Antagonist IL-18 Binding Protein Is Produced in the Human
Author Manuscript Published OnlineFirst on July 19, 2013; DOI: 10.1158/1078-0432.CCR-13-0568 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. The IL-18 antagonist IL-18 Binding Protein is produced in the human ovarian cancer microenvironment Grazia Carbotti1§, Gaia Barisione1, Anna Maria Orengo1, Antonella Brizzolara1, Irma Airoldi2, Marina Bagnoli3, Patrizia Pinciroli3, Delia Mezzanzanica3, Maria Grazia Centurioni4, Marina Fabbi1*, Silvano Ferrini1* 1Department of Integrated Oncological Therapies, IRCCS AOU San Martino-IST Istituto Nazionale per la Ricerca sul Cancro, Genoa, Italy; 2Laboratory of Oncology, IRCCS Istituto G. Gaslini, Genoa Italy; 3Department of Experimental Oncology and Molecular Medicine, Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, Italy; 4Department of Surgery, IRCCS AOU San Martino-IST Istituto Nazionale per la Ricerca sul Cancro, Genoa, Italy. Running title: IL-18BP in ovarian cancer Key words: IL-18 Binding Protein, ovarian cancer, immune escape, IL-27, EBI3 Financial support: AIRC (Associazione Italiana per la Ricerca sul Cancro, IG5509, IG13018 and IG13518), Ministry of Health Project ‘Tumori Femminili’ and Compagnia di San Paolo. §Grazia Carbotti is enrolled in the Doctorate School of Genetics, University of Genoa, Italy. *Equal contributors and correspondence: Silvano Ferrini and Marina Fabbi, UOC Terapia Immunologica, IRCCS AOU San Martino-IST Istituto Nazionale per la Ricerca sul Cancro, Largo R. Benzi 10, 16132 Genova, ITALY Phone: 0039-010-5737-372; Fax:-374; Email: [email protected], [email protected] Conflict of interest: no conflict of interest to disclose. Word count of text: 4667 Total number of figures and tables: 6 Relevance Statement: IL-18 is an immune-enhancing cytokine, which is being studied in clinical trials of immunotherapy.