Characterization of Differential Dynamics, Specificity, and Allostery
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Interactions Between the Parasite Philasterides Dicentrarchi and the Immune System of the Turbot Scophthalmus Maximus.A Transcriptomic Analysis
biology Article Interactions between the Parasite Philasterides dicentrarchi and the Immune System of the Turbot Scophthalmus maximus.A Transcriptomic Analysis Alejandra Valle 1 , José Manuel Leiro 2 , Patricia Pereiro 3 , Antonio Figueras 3 , Beatriz Novoa 3, Ron P. H. Dirks 4 and Jesús Lamas 1,* 1 Department of Fundamental Biology, Institute of Aquaculture, Campus Vida, University of Santiago de Compostela, 15782 Santiago de Compostela, Spain; [email protected] 2 Department of Microbiology and Parasitology, Laboratory of Parasitology, Institute of Research on Chemical and Biological Analysis, Campus Vida, University of Santiago de Compostela, 15782 Santiago de Compostela, Spain; [email protected] 3 Institute of Marine Research, Consejo Superior de Investigaciones Científicas-CSIC, 36208 Vigo, Spain; [email protected] (P.P.); antoniofi[email protected] (A.F.); [email protected] (B.N.) 4 Future Genomics Technologies, Leiden BioScience Park, 2333 BE Leiden, The Netherlands; [email protected] * Correspondence: [email protected]; Tel.: +34-88-181-6951; Fax: +34-88-159-6904 Received: 4 September 2020; Accepted: 14 October 2020; Published: 15 October 2020 Simple Summary: Philasterides dicentrarchi is a free-living ciliate that causes high mortality in marine cultured fish, particularly flatfish, and in fish kept in aquaria. At present, there is still no clear picture of what makes this ciliate a fish pathogen and what makes fish resistant to this ciliate. In the present study, we used transcriptomic techniques to evaluate the interactions between P. dicentrarchi and turbot leucocytes during the early stages of infection. The findings enabled us to identify some parasite genes/proteins that may be involved in virulence and host resistance, some of which may be good candidates for inclusion in fish vaccines. -

The Role of Ferroptosis in Breast Cancer Patients: a Comprehensive Analysis Zeng-Hong Wu1,2, Yun Tang3,Hongyu1 and Hua-Dong Li4
Wu et al. Cell Death Discovery (2021) 7:93 https://doi.org/10.1038/s41420-021-00473-5 Cell Death Discovery ARTICLE Open Access The role of ferroptosis in breast cancer patients: a comprehensive analysis Zeng-Hong Wu1,2, Yun Tang3,HongYu1 and Hua-Dong Li4 Abstract Breast cancer (BC) affects the breast tissue and is the second most common cause of mortalities among women. Ferroptosis is an iron-dependent cell death mode that is characterized by intracellular accumulation of reactive oxygenspecies(ROS).Weconstructed a prognostic multigene signature based on ferroptosis-associated differentially expressed genes (DEGs). Moreover, we comprehensively analyzed the role of ferroptosis-associated miRNAs, lncRNAs, and immune responses. A total of 259 ferroptosis-related genes wereextracted.KEGGfunction analysis of these genes revealed that they were mainly enriched in the HIF-1 signaling pathway, NOD-like receptor signaling pathway, central carbon metabolism in cancer, and PPAR signaling pathway. Fifteen differentially expressed genes (ALOX15, ALOX15B, ANO6, BRD4, CISD1, DRD5, FLT3, G6PD, IFNG, NGB, NOS2, PROM2, SLC1A4, SLC38A1,andTP63) were selected as independent prognostic factors for BC patients. Moreover, T cell functions, including the CCR score, immune checkpoint, cytolytic activity, HLA, inflammation promotion, para-inflammation, T cell co-stimulation, T cell co-inhibition, and type II INF responses were significantly different between the low- risk and high-risk groups of the TCGA cohort. Immune checkpoints between the two groups revealed that the expressions of PDCD-1 (PD-1), CTLA4, LAG3, TNFSF4/14, TNFRSF4/8/9/14/18/25,andIDO1/2 among others were significantly different. A total of 1185 ferroptosis-related lncRNAs and 219 ferroptosis-related miRNAs were also included in this study. -

Quantitative Proteomics Analysis of Young and Elderly Skin with DIA Mass Spectrometry Reveals New Skin Aging-Related Proteins
www.aging-us.com AGING 2020, Vol. 12, No. 13 Research Paper Quantitative proteomics analysis of young and elderly skin with DIA mass spectrometry reveals new skin aging-related proteins Jing Ma1,2, Mengting Liu1,2, Yaochi Wang1,2, Cong Xin1,2, Hui Zhang1,2, Shirui Chen1,2, Xiaodong Zheng1,2, Xuejun Zhang1,2, Fengli Xiao1,2,3, Sen Yang1,2 1Department of Dermatology of First Affiliated Hospital, and Institute of Dermatology, Anhui Medical University, Hefei, Anhui, China 2Key Laboratory of Dermatology, Anhui Medical University, Ministry of Education, Hefei, Anhui, China 3The Center for Scientific Research of Anhui Medical University, Hefei, Anhui, China Correspondence to: Fengli Xiao, Sen Yang; email: [email protected], [email protected] Keywords: aging, epidermal proteins, skin rejuvenation and aging, proteome, mass spectrometer Received: February 25, 2020 Accepted: May 27, 2020 Published: June 29, 2020 Copyright: Ma et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Skin aging is a specific manifestation of the physiological aging process that occurs in virtually all organisms. In this study, we used data independent acquisition mass spectrometry to perform a comparative analysis of protein expression in volar forearm skin samples from of 20 healthy young and elderly Chinese individuals. Our quantitative proteomic analysis identified a total of 95 differentially expressed proteins (DEPs) in aged skin compared to young skin. Enrichment analyses of these DEPs (57 upregulated and 38 downregulated proteins) based on the GO, KEGG, and KOG databases revealed functional clusters associated with immunity and inflammation, oxidative stress, biosynthesis and metabolism, proteases, cell proliferation, cell differentiation, and apoptosis. -

(ALOX15B) (NM 001039130) Human Tagged ORF Clone Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC214018 15 Lipoxygenase 2 (ALOX15B) (NM_001039130) Human Tagged ORF Clone Product data: Product Type: Expression Plasmids Product Name: 15 Lipoxygenase 2 (ALOX15B) (NM_001039130) Human Tagged ORF Clone Tag: Myc-DDK Symbol: ALOX15B Synonyms: 15-LOX-2 Vector: pCMV6-Entry (PS100001) E. coli Selection: Kanamycin (25 ug/mL) Cell Selection: Neomycin This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 5 15 Lipoxygenase 2 (ALOX15B) (NM_001039130) Human Tagged ORF Clone – RC214018 ORF Nucleotide >RC214018 representing NM_001039130 Sequence: Red=Cloning site Blue=ORF Green=Tags(s) TTTTGTAATACGACTCACTATAGGGCGGCCGGGAATTCGTCGACTGGATCCGGTACCGAGGAGATCTGCC GCCGCGATCGCC ATGGCCGAGTTCAGGGTCAGGGTGTCCACCGGAGAAGCCTTCGGGGCTGGCACATGGGACAAAGTGTCTG TCAGCATCGTGGGGACCCGGGGAGAGAGCCCCCCACTGCCCCTGGACAATCTCGGCAAGGAGTTCACTGC GGGCGCTGAGGAGGACTTCCAGGTGACGCTCCCGGAGGACGTAGGCCGAGTGCTGCTGCTGCGCGTGCAC AAGGCGCCCCCAGTGCTGCCCCTGCTGGGGCCCCTGGCCCCGGATGCCTGGTTCTGCCGCTGGTTCCAGC TGACACCGCCGCGGGGCGGCCACCTCCTCTTCCCCTGCTACCAGTGGCTGGAGGGGGCGGGGACCCTGGT GCTGCAGGAGGGTACAGCCAAGGTGTCCTGGGCAGACCACCACCCTGTGCTCCAGCAACAGCGCCAGGAG GAGCTTCAGGCCCGGCAGGAGATGTACCAGTGGAAGGCTTACAACCCAGGTTGGCCTCACTGCCTGGATG AAAAGACAGTGGAAGACTTGGAGCTCAATATCAAATACTCCACAGCCAAGAATGCCAACTTTTATCTACA -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Electronic Supplementary Material (ESI) for Metallomics
Electronic Supplementary Material (ESI) for Metallomics. This journal is © The Royal Society of Chemistry 2018 Uniprot Entry name Gene names Protein names Predicted Pattern Number of Iron role EC number Subcellular Membrane Involvement in disease Gene ontology (biological process) Id iron ions location associated 1 P46952 3HAO_HUMAN HAAO 3-hydroxyanthranilate 3,4- H47-E53-H91 1 Fe cation Catalytic 1.13.11.6 Cytoplasm No NAD biosynthetic process [GO:0009435]; neuron cellular homeostasis dioxygenase (EC 1.13.11.6) (3- [GO:0070050]; quinolinate biosynthetic process [GO:0019805]; response to hydroxyanthranilate oxygenase) cadmium ion [GO:0046686]; response to zinc ion [GO:0010043]; tryptophan (3-HAO) (3-hydroxyanthranilic catabolic process [GO:0006569] acid dioxygenase) (HAD) 2 O00767 ACOD_HUMAN SCD Acyl-CoA desaturase (EC H120-H125-H157-H161; 2 Fe cations Catalytic 1.14.19.1 Endoplasmic Yes long-chain fatty-acyl-CoA biosynthetic process [GO:0035338]; unsaturated fatty 1.14.19.1) (Delta(9)-desaturase) H160-H269-H298-H302 reticulum acid biosynthetic process [GO:0006636] (Delta-9 desaturase) (Fatty acid desaturase) (Stearoyl-CoA desaturase) (hSCD1) 3 Q6ZNF0 ACP7_HUMAN ACP7 PAPL PAPL1 Acid phosphatase type 7 (EC D141-D170-Y173-H335 1 Fe cation Catalytic 3.1.3.2 Extracellular No 3.1.3.2) (Purple acid space phosphatase long form) 4 Q96SZ5 AEDO_HUMAN ADO C10orf22 2-aminoethanethiol dioxygenase H112-H114-H193 1 Fe cation Catalytic 1.13.11.19 Unknown No oxidation-reduction process [GO:0055114]; sulfur amino acid catabolic process (EC 1.13.11.19) (Cysteamine -

Reduced 15-Lipoxygenase 2 and Lipoxin A4/Leukotriene B4 Ratio in Children with Cystic Fibrosis
ORIGINAL ARTICLE CYSTIC FIBROSIS Reduced 15-lipoxygenase 2 and lipoxin A4/leukotriene B4 ratio in children with cystic fibrosis Fiona C. Ringholz1, Paul J. Buchanan1, Donna T. Clarke1, Roisin G. Millar1, Michael McDermott2, Barry Linnane1,3,4, Brian J. Harvey5, Paul McNally1,2 and Valerie Urbach1,6 Affiliations: 1National Children’s Research Centre, Crumlin, Dublin, Ireland. 2Our Lady’s Children’s Hospital, Crumlin, Dublin, Ireland. 3Midwestern Regional Hospital, Limerick, Ireland. 4Centre for Interventions in Infection, Inflammation and Immunity (4i), Graduate Entry Medical School, University of Limerick, Limerick, Ireland. 5Molecular Medicine Laboratories, Royal College of Surgeons in Ireland, Beaumont Hospital, Dublin, Ireland. 6Institut National de la Sante´ et de la Recherche Me´dicale, U845, Faculte´ de Me´decine Paris Descartes, Paris, France. Correspondence: Valerie Urbach, National Children’s Research Centre, Crumlin, Dublin 12, Ireland. E-mail: [email protected] ABSTRACT Airway disease in cystic fibrosis (CF) is characterised by impaired mucociliary clearance, persistent bacterial infection and neutrophilic inflammation. Lipoxin A4 (LXA4) initiates the active resolution of inflammation and promotes airway surface hydration in CF models. 15-Lipoxygenase (LO) plays a central role in the ‘‘class switch’’ of eicosanoid mediator biosynthesis from leukotrienes to lipoxins, initiating the active resolution of inflammation. We hypothesised that defective eicosanoid mediator class switching contributes to the failure to resolve inflammation in CF lung disease. Using bronchoalveolar lavage (BAL) samples from 46 children with CF and 19 paediatric controls we demonstrate that the ratio of LXA4 to leukotriene B4 (LTB4) is depressed in CF BAL (p,0.01), even in the absence of infection (p,0.001). -
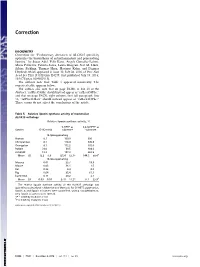
Evolutionary Alteration of ALOX15 Specificity Optimizes The
Correction BIOCHEMISTRY Correction for “Evolutionary alteration of ALOX15 specificity optimizes the biosynthesis of antiinflammatory and proresolving lipoxins,” by Susan Adel, Felix Karst, Àngels González-Lafont, Mária Pekárová, Patricia Saura, Laura Masgrau, José M. Lluch, Sabine Stehling, Thomas Horn, Hartmut Kuhn, and Dagmar Heydeck, which appeared in issue 30, July 26, 2016, of Proc Natl Acad Sci USA (113:E4266–E4275; first published July 13, 2016; 10.1073/pnas.1604029113). The authors note that Table 5 appeared incorrectly. The corrected table appears below. The authors also note that on page E4266, in line 20 of the Abstract, “ratPhe353Ala” should instead appear as “ratLeu353Phe;” and that on page E4270, right column, first full paragraph, line 12, “ratPhe353Leu” should instead appear as “ratLeu353Phe.” These errors do not affect the conclusions of the article. Table 5. Relative lipoxin synthase activity of mammalian ALOX15 orthologs Relative lipoxin synthase activity, % 5-HETE as 5,6-DiHETE as Species 15-/12-ratio substrate substrate 15-lipoxygenating Human 8.1 100.0 100 Chimpanzee 8.1 118.0 145.8 Orangutan 8.1 172.2 105.6 Rabbit 24.0 39.5 108.6 ratL353F 13.3 197.3 262.5 Mean ± SD 12.3 ± 6.9 125.4 ± 62.1* 144.5 ± 68.4† 12-lipoxygenating Macaca 0.01 25.7 19.9 Mouse 0.03 36.1 1.5 Rat 0.26 8.4 0.0 Pig 0.04 35.4 61.1 humI418A 0.11 29.2 2.1 Mean ± SD 0.09 ± 0.10 27.0 ± 11.2* 17.1 ± 25.9† The relative lipoxin synthase activity of the ALOX15 orthologs was quantified as described in Materials and Methods. -

Supplementary Table 1: List of the 316 Genes Regulated During Hyperglycemic Euinsulinemic Clamp in Skeletal Muscle
Supplementary Table 1: List of the 316 genes regulated during hyperglycemic euinsulinemic clamp in skeletal muscle. UGCluster Name Symbol Fold Change Cytoband Response to stress Hs.517581 Heme oxygenase (decycling) 1 HMOX1 3.80 22q12 Hs.374950 Metallothionein 1X MT1X 2.20 16q13 Hs.460867 Metallothionein 1B (functional) MT1B 1.70 16q13 Hs.148778 Oxidation resistance 1 OXR1 1.60 8q23 Hs.513626 Metallothionein 1F (functional) MT1F 1.47 16q13 Hs.534330 Metallothionein 2A MT2A 1.45 16q13 Hs.438462 Metallothionein 1H MT1H 1.42 16q13 Hs.523836 Glutathione S-transferase pi GSTP1 -1.74 11q13 Hs.459952 Stannin SNN -1.92 16p13 Immune response, cytokines & related Hs.478275 TNF (ligand) superfamily, member 10 (TRAIL) TNFSF10 1.58 3q26 Hs.278573 CD59 antigen p18-20 (protectin) CD59 1.49 11p13 Hs.534847 Complement component 4B, telomeric C4A 1.47 6p21.3 Hs.535668 Immunoglobulin lambda variable 6-57 IGLV6-57 1.40 22q11.2 Hs.529846 Calcium modulating ligand CAMLG -1.40 5q23 Hs.193516 B-cell CLL/lymphoma 10 BCL10 -1.40 1p22 Hs.840 Indoleamine-pyrrole 2,3 dioxygenase INDO -1.40 8p12-p11 Hs.201083 Mal, T-cell differentiation protein 2 MAL2 -1.44 Hs.522805 CD99 antigen-like 2 CD99L2 -1.45 Xq28 Hs.50002 Chemokine (C-C motif) ligand 19 CCL19 -1.45 9p13 Hs.350268 Interferon regulatory factor 2 binding protein 2 IRF2BP2 -1.47 1q42.3 Hs.567249 Contactin 1 CNTN1 -1.47 12q11-q12 Hs.132807 MHC class I mRNA fragment 3.8-1 3.8-1 -1.48 6p21.3 Hs.416925 Carcinoembryonic antigen-related cell adhesion molecule 19 CEACAM19 -1.49 19q13.31 Hs.89546 Selectin E (endothelial -

Functional Characterization of Genetic Enzyme Variations in Human Lipoxygenases
Redox Biology 1 (2013) 566–577 Contents lists available at ScienceDirect Redox Biology journal homepage: www.elsevier.com/locate/redox Functional characterization of genetic enzyme variations in human lipoxygenases Thomas Horn a,n, Kumar Reddy Kakularam b, Monika Anton a, Constanze Richter c, Pallu Reddanna b,d, Hartmut Kuhn a a Institute of Biochemistry, University Medicine Berlin—Charité, Charitéplatz 1, D-10117 Berlin, Germany b Department of Animal Sciences, School of Life Sciences, University of Hyderabad, Gachibowli, Hyderabad 500046, Andhra Pradesh, India c Institute of Nutrition Technology and Nutrition Chemistry, TU Berlin, Gustav-Meyer-Allee 25, D-13355 Berlin, Germany d National Institute of Animal Biotechnology, Hyderabad 500046, Andhra Pradesh, India article info abstract Article history: Mammalian lipoxygenases play a role in normal cell development and differentiation but they have also been Received 28 October 2013 implicated in the pathogenesis of cardiovascular, hyperproliferative and neurodegenerative diseases. As lipid Accepted 1 November 2013 peroxidizing enzymes they are involved in the regulation of cellular redox homeostasis since they produce lipid hydroperoxides, which serve as an efficient source for free radicals. There are various epidemiological Keywords: correlation studies relating naturally occurring variationsinthesixhumanlipoxygenasegenes(SNPsorrare Eicosanoids mutations) to the frequency for various diseases in these individuals, but for most of the described variations no Leukotrienes functional data are available. Employing a combined bioinformatical and enzymological strategy, which Lipoxygenases included structural modeling and experimental site-directed mutagenesis, we systematically explored the Gene polymorphism structural and functional consequences of non-synonymous genetic variations in four different human SNP lipoxygenase genes (ALOX5, ALOX12, ALOX15, and ALOX15B) that have been identified in the human 1000 genome project. -

Human ALOX15B / 15 Lipoxygenase 2 Protein
Human ALOX15B / 15 Lipoxygenase 2 Protein Catalog Number: 14564-HNCB General Information SDS-PAGE: Gene Name Synonym: 15-LOX-2; ALOX15B Protein Construction: A DNA sequence encoding the human ALOX15B (AAH35217.1)( Met1- Ile676) was expressed and purified with two additional amino acids (Gly & Pro ) at the N-terminus. Source: Human Expression Host: Baculovirus-Insect Cells QC Testing Purity: > 90 % as determined by SDS-PAGE Endotoxin: Protein Description < 1.0 EU per μg of the protein as determined by the LAL method ALOX15B is a member of the lipoxygenase family of structurally related nonheme iron dioxygenases involved in the production of fatty acid Stability: hydroperoxides. ALOX15B converts arachidonic acid exclusively to 15S- hydroperoxyeicosatetraenoic acid, while metabolizing linoleic acid less Samples are stable for up to twelve months from date of receipt at -70 ℃ effectively. ALOX15B gene is located in a cluster of related genes and a pseudogene that spans approximately 100 kilobases on the short arm of Predicted N terminal: Gly chromosome 17. Alternatively spliced transcript variants encoding different Molecular Mass: isoforms have been described. The secreted recombinant human ALOX15B consists of 678 amino acids References and predicts a molecular mass of 76 KDa. The apparent molecular mass 1.Kilty I. et al., 2000, Eur J Biochem. 266 (1): 83-93. 2.Sigal E. et al., 1990, of the protein is approximately 62-66 KDa in SDS-PAGE under reducing J Biol Chem. 265 (9): 5113-20. 3.Brash AR. et al., 1997, Proc Natl Acad conditions due to glycosylation. Sci. 94 (12): 6148-52. Formulation: Lyophilized from sterile 20mM Tris, 500mM NaCl, 10% glycerol, pH 7.4. -

Sex-Dependent Gene Expression After Ochratoxin a Insult in F344 Rat
Food and Chemical Toxicology 123 (2019) 337–348 Contents lists available at ScienceDirect Food and Chemical Toxicology journal homepage: www.elsevier.com/locate/foodchemtox Sex-dependent gene expression after ochratoxin A insult in F344 rat kidney T ∗ Ariane Vettorazzia,b, ,1, Laura Pastora,1, Elizabeth Guruceagab,c, Adela López de Ceraina,b a University of Navarra, Department of Pharmacology and Toxicology, Faculty of Pharmacy and Nutrition, E-31008, Pamplona, Spain b IdiSNA, Navarra Institute for Health Research, E-31008, Pamplona, Spain c Bioinformatics Platform, Center for Applied Medical Research (CIMA), University of Navarra, E-31008, Pamplona, Spain ARTICLE INFO ABSTRACT Keywords: Ochratoxin A (OTA) is a potent rodent nephrocarcinogen; being males more sensitive than females. The ob- Ochratoxin A (OTA) jective was to study the response between sexes at gene expression level (whole genome transcriptomics) in Sex differences kidneys of F344 rats treated with 0.21 or 0.50 mg/kg bw OTA for 21 days. DNA methylation analysis of selected Gene expression genes was also studied (MALDI-TOF mass spectrometry). OTA-induced response was dose-dependent in males Metabolism and females, although clearer in males. Females showed a higher number of altered genes than males but Nuclear receptors functional analysis revealed a higher number of significantly enriched toxicity lists in 0.21 mg/kg treated males. Methylation OTA modulated damage, signaling and metabolism related lists, as well as inflammation, proliferation and oxidative stress in both sexes. Eleven toxicity lists (damage, fibrosis, cell signaling and metabolism) were ex- clusively altered in males while renal safety biomarker and biogenesis of mitochondria lists were exclusively enriched in females.