The Management of Sickle Cell Disease
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Research Training in the Biomedical, Behavioral and Clinical Sciences
RESEARCH TRAINING IN THE BIOMEDICAL, BEHAVIORAL, AND CLINICAL RESEARCH SCIENCES Committee to Study the National Needs for Biomedical, Behavioral, and Clinical Research Personnel Board on Higher Education and Workforce Policy and Global Affairs THE NATIONAL ACADEMIES PRESS 500 Fifth Street, N.W. Washington, DC 20001 NOTICE: The project that is the subject of this report was approved by the Governing Board of the National Research Council, whose members are drawn from the councils of the National Academy of Sciences, the National Academy of Engineering, and the Institute of Medicine. The members of the committee responsible for the report were chosen for their special competences and with regard for appropriate balance. This project was supported by Contract/Grant No. DHHS-5294, Task Order #187 between the National Acad- emy of Science and the National Institutes of Health, Department of Health and Human Services. Any opinions, findings, conclusions, or recommendations expressed in this publication are those of the Committee to Study the National Needs for Biomedical, Behavioral, and Clinical Research Personnel and do not necessarily reflect the views of the organizations or agencies that provided support for the project. International Standard Book Number-13: 978-0-309-15965-4 (Book) International Standard Book Number-10: 0-309-15965-2 (Book) International Standard Book Number-13: 978-0-309-15966-1 (PDF) International Standard Book Number-10: 0-309-15966-0 (PDF) Library of Congress Control Number: 2011921184 Additional copies of this report are available from The National Academies Press, 500 Fifth Street, N.W., Lockbox 285, Washington, DC 20055; (800) 624-6242 or (202) 334-3313 (in the Washington metropolitan area); Internet, http://www.nap.edu. -

Hemolysis and Venous Thrombosis: Which Link? A
ISSN: 2378-3656 Rkiouak et al. Clin Med Rev Case Rep 2020, 7:329 DOI: 10.23937/2378-3656/1410329 Volume 7 | Issue 11 Clinical Medical Reviews Open Access and Case Reports ORIGINAL RESEARCH Hemolysis and Venous Thrombosis: Which Link? A. Rkiouak, PH.D1, I El Kassimi, MD2, N Sahel, MD2, M Zaizaa, MD2 and Y Sekkach, PhD1 Check for updates Internal Medicine A Department, Mohammed V Military Hospital Medical School of Rabat, Morocco *Corresponding author: Adil Rkiouak, PH.D., Department of Internal Medicine A, Mohammed V Military Hospital, Medical School of Rabat, Morocco, Tel: +212-66-179-44-04 The mechanism of antibody-mediated hemolysis is Abstract via phagocytosis or complement-mediated destruction The association hemolysis and venous thrombosis remains and can occur intravascular or extravascular. The intra- unknown to clinicians, despite our advances in comrehen- sion of pathophysiological bases. vascular mechanisms include direct cellular destruction via lysis, toxins, or trauma; fragmentation and oxida- Haemolysis, which is observed in multiple diseases, can affect all three components of Virchow’s triad. It is not sur- tion. prising that there is a link between haemolytic disorders and Multiple haemolytic disorders produce substantial thrombosis. intravascular haemolysis. Examples, the corpuscular We will try to clarify the main pro-thrombotic mechanisms hemolysis include PNH, extra-corpuscular haemolysis, during hemolysis through 3 clinical observations of deep ve- acquired (autoimmune haemolytic anaemia (AIHA , nous thrombosis in 3 main types of hemolytic pathologies, ) namely a case of paroxysmal nocturnal hemoglobinuria, thrombotic thrombocytopenic purpura (PTT)), as well thrombotic thrombocytopenic purpura and autoimmune as other diseases. These disorders are also associated anemia hemolytic. -

Clostridial Septicemia with Intravascular Hemolysis: a Case Report G
Henry Ford Hospital Medical Journal Volume 13 | Number 4 Article 4 12-1965 Clostridial Septicemia With Intravascular Hemolysis: A Case Report G. M. Mastio E. Morfin Follow this and additional works at: https://scholarlycommons.henryford.com/hfhmedjournal Part of the Life Sciences Commons, Medical Specialties Commons, and the Public Health Commons Recommended Citation Mastio, G. M. and Morfin, E. (1965) "Clostridial Septicemia With Intravascular Hemolysis: A Case Report," Henry Ford Hospital Medical Bulletin : Vol. 13 : No. 4 , 421-425. Available at: https://scholarlycommons.henryford.com/hfhmedjournal/vol13/iss4/4 This Article is brought to you for free and open access by Henry Ford Health System Scholarly Commons. It has been accepted for inclusion in Henry Ford Hospital Medical Journal by an authorized editor of Henry Ford Health System Scholarly Commons. Henry Ford Hosp. Med. Bull. Vol. 13, December 1965 CLOSTRIDIAL SEPTICEMIA WITH INTRAVASCULAR HEMOLYSIS A CASE REPORT G. M. MASTIC, M.D. AND E. MORFIN, M.D. In 1871 Bottini' demonstrated the bacterial nature of gas gangrene, but failed to isolate a causal organism. Clostridium perfringens, sometimes known as Clostridium welchii, was discovered independently during 1892 and 1893 by Welch, Frankel, "Veillon and Zuber.^ This organism is a saprophytic inhabitant of the intestinal tract, and may be a harmless saprophyte of the female genital tract occurring in the vagina in 4-6 per cent of pregnant women. Clostridial organisms occur in great numbers and distribution throughout the world. Because of this, they are very common in traumatic wounds. Very few species of Clostridia, however, are pathogenic, and still fewer are capable of producing gas gangrene in man. -

Diagnosis of Sickle Cell Disease and HBB Haplotyping in the Era of Personalized Medicine: Role of Next Generation Sequencing
Journal of Personalized Medicine Article Diagnosis of Sickle Cell Disease and HBB Haplotyping in the Era of Personalized Medicine: Role of Next Generation Sequencing Adekunle Adekile 1,*, Nagihan Akbulut-Jeradi 2, Rasha Al Khaldi 2, Maria Jinky Fernandez 2 and Jalaja Sukumaran 1 1 Department of Pediatrics, Faculty of Medicine, Kuwait University, P.O. Box 24923, Safat 13110, Kuwait; jalajasukumaran@hotmail 2 Advanced Technology Company, Hawali 32060, Kuwait; [email protected] (N.A.-J.); [email protected] (R.A.); [email protected] (M.J.F.) * Correspondence: [email protected]; Tel.: +965-253-194-86 Abstract: Hemoglobin genotype and HBB haplotype are established genetic factors that modify the clinical phenotype in sickle cell disease (SCD). Current methods of establishing these two factors are cumbersome and/or prone to errors. The throughput capability of next generation sequencing (NGS) makes it ideal for simultaneous interrogation of the many genes of interest in SCD. This study was designed to confirm the diagnosis in patients with HbSS and Sβ-thalassemia, identify any ß-thal mutations and simultaneously determine the ßS HBB haplotype. Illumina Ampliseq custom DNA panel was used to genotype the DNA samples. Haplotyping was based on the alleles on five haplotype-specific SNPs. The patients studied included 159 HbSS patients and 68 Sβ-thal patients, previously diagnosed using high performance liquid chromatography (HPLC). There was Citation: Adekile, A.; considerable discordance between HPLC and NGS results, giving a false +ve rate of 20.5% with a S Akbulut-Jeradi, N.; Al Khaldi, R.; sensitivity of 79% for the identification of Sβthal. -
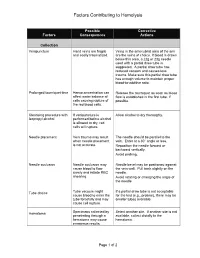
Factors Contributing to Hemolysis
Factors Contributing to Hemolysis Possible Corrective Factors Consequences Actions Collection Venipuncture Hand veins are fragile Veins in the antecubital area of the arm and easily traumatized. are the veins of choice. If blood is drawn below this area, a 22g or 23g needle used with a partial draw tube is suggested. A partial draw tube has reduced vacuum and causes less trauma. Make sure this partial draw tube has enough volume to maintain proper blood-to-additive ratio. Prolonged tourniquet time Hemoconcentration can Release the tourniquet as soon as blood affect water balance of flow is established in the first tube, if cells causing rupture of possible. the red blood cells. Cleansing procedure with If venipuncture is Allow alcohol to dry thoroughly. isopropyl alcohol performed before alcohol is allowed to dry, red cells will rupture. Needle placement Vein trauma may result The needle should be parallel to the when needle placement vein. Enter at a 30° angle or less. is not accurate. Reposition the needle forward or backward vertically. Avoid probing. Needle occlusion Needle occlusion may Needle bevel may be positioned against cause blood to flow the vein wall. Pull back slightly on the slowly and initiate RBC needle. shearing. Avoid rotating or changing the angle of the needle. Tube choice Tube vacuum might If a partial draw tube is not acceptable cause blood to enter the for the test (e.g., protime), there may be tube forcefully and may smaller tubes available. cause cell rupture. Hematoma Specimens collected by Select another site. If another site is not penetrating through a available, collect distally to the hematoma may cause hematoma. -

PATHOLOGY RESIDENT HEMATOLOGY ROTATION (North Florida/South Georgia Veterans Health Care System): Rotation Director: William L
PATHOLOGY RESIDENT HEMATOLOGY ROTATION (North Florida/South Georgia Veterans Health Care System): Rotation Director: William L. Clapp, M.D., Chief, Hematology Section, Gainesville VAMC; Consultants: Neil S. Harris, M.D., Director, Laboratory Hematology/Coagulation, University of Florida and Shands Hospital and Raul C. Braylan, M.D., Director, Hematopathology, University of Florida and Shands Hospital 1. Description of the Rotation: In this rotation, the resident will gain experience in laboratory hematology, which will include (1) peripheral blood studies to evaluate a variety of hematologic disorders, including anemias, lymphoproliferative and myeloproliferative disorders and leukemias. The emphasis on a multidisciplinary approach to diagnose hematologic disorders (including correlation of the peripheral blood studies with bone marrow and lymph node studies) provides an opportunity for the resident to also gain additional experience in (2) traditional histopathology, (3) immunohistochemistry, (4) electron microscopy, (5) protein electrophoresis, (6) flow cytometry, (7) cytogenetics and (8) molecular genetics which may be performed on the peripheral blood, bone marrow or lymph nodes of patients. The residents will acquire valuable experience by independently performing some bone marrow procedures. In addition, the resident will gain experience in coagulation testing. The residents will become familiar with the instrumentation in the hematology laboratory, including the operating principles and trouble-shooting (medical knowledge). The availability of assembled case study sets and reading materials (medical knowledge) will enhance the resident’s experience. Participation in CAP surveys, continuing education and hematology conferences is a component of the rotation (practice-based learning). Management issues and computer applications will be discussed (practice-based learning). As appropriate to the individual case or consultation under review, the ethical, socioeconomic, medicolegal and cost-containment issues will be reviewed and discussed. -

Thalassemia, Hemophilia & Sickle Cell Disease
10/22/2018 Global Best Practices in Care, Rehabilitation and Research Thalassemia Syndrome Blood Disorders (Thalassemia, Hemophilia & Sickle Cell Disease) Dr. J.S. Arora Thalassemialogist 7% of the world population MSc in Haemoglobinopathy University College London Carry Thalassemia/Hb’pathy gene General secretary: National Thalassemia Welfare Society Federation of Indian Thalassemics Member Ethics Committee: 400 million heterozygous carriers IIT Delhi Lady Hardinge Medical College and Associated Hospitals, New Delhi ITS Dental College Hospital & Research Centre, Greater NOIDA 3,00,000-4,00,000 babies with severe Founder Member: Indian Alliance of Patient Groups haemoglobinopathies born each year. Founding Trustee: Genomics And Public Health Foundation Formerly Coordinator Thalassemia Cell Govt. of Delhi Member Advisory Committee - D D U Hospital Govt. of Delhi INDIA , THAILAND AND INDONESIA “Life Time Service Award” from PHO Chambers of IAP ► 50% OF WORLD’S THALASSAEMIA CARRIERS Patients for Patient Safety (PFPS) Champion India Member: Patients for Patient Safety Advisory Group ► 50% OF THALASSAEMIA MAJORS [email protected] β Thalassemia Who are affected 100 million carriers of β Thalassemia. More than 100,000 Thalassemia Major born/year India • thalassemia : Carrier rate 1% - 17% (mean 3.9%) more prevalent in certain communities. 50 million carriers, Over 12,000 affected born every year. • Sickle Cell Disease : common in tribes carrier rate as high as 40% in some areas • HbE : Highly prevalent in West Bengal & North Eastern -

Section 8: Hematology CHAPTER 47: ANEMIA
Section 8: Hematology CHAPTER 47: ANEMIA Q.1. A 56-year-old man presents with symptoms of severe dyspnea on exertion and fatigue. His laboratory values are as follows: Hemoglobin 6.0 g/dL (normal: 12–15 g/dL) Hematocrit 18% (normal: 36%–46%) RBC count 2 million/L (normal: 4–5.2 million/L) Reticulocyte count 3% (normal: 0.5%–1.5%) Which of the following caused this man’s anemia? A. Decreased red cell production B. Increased red cell destruction C. Acute blood loss (hemorrhage) D. There is insufficient information to make a determination Answer: A. This man presents with anemia and an elevated reticulocyte count which seems to suggest a hemolytic process. His reticulocyte count, however, has not been corrected for the degree of anemia he displays. This can be done by calculating his corrected reticulocyte count ([3% × (18%/45%)] = 1.2%), which is less than 2 and thus suggestive of a hypoproliferative process (decreased red cell production). Q.2. A 25-year-old man with pancytopenia undergoes bone marrow aspiration and biopsy, which reveals profound hypocellularity and virtual absence of hematopoietic cells. Cytogenetic analysis of the bone marrow does not reveal any abnormalities. Despite red blood cell and platelet transfusions, his pancytopenia worsens. Histocompatibility testing of his only sister fails to reveal a match. What would be the most appropriate course of therapy? A. Antithymocyte globulin, cyclosporine, and prednisone B. Prednisone alone C. Supportive therapy with chronic blood and platelet transfusions only D. Methotrexate and prednisone E. Bone marrow transplant Answer: A. Although supportive care with transfusions is necessary for treating this patient with aplastic anemia, most cases are not self-limited. -

BPS White Paper
BOARD OF PHARMACY SPECIALTIES WHITE PAPER Five‐Year Vision for Pharmacy Specialties Approved by the BPS Board of Directors on January 12, 2013 © Copyright 2013, Board of Pharmacy Specialties Introduction The Joint Commission of Pharmacy Practitioners’ (JCPP) Future Vision of Pharmacy Practice 2015 states, “Pharmacists will be the healthcare professionals responsible for providing patient care that ensures optimal medication therapy outcomes.” i As the scope of pharmacy practice evolves to meet the complex medication‐related needs of patients, board certification is critical to assure stakeholders of the level of knowledge and skills of pharmacists who provide direct patient care. The Board of Pharmacy Specialties (BPS) acknowledges the support and collaboration of many national pharmacy organizations in this pursuit and assumes the responsibility for recognition of pharmacy specialization through board certification.ii,iii, iv In assuming this responsibility, BPS also acknowledges the pharmacist’s role in direct patient care as it continues to evolve against a backdrop of many unknowns such as changing health care delivery systems and payment models in which the role of government and the private sector are in a state of flux. Whatever changes come to fruition in the delivery of health care, BPS must move forward in a flexible manner that meets its mission to improve patient care by promoting the recognition and value of specialized training, knowledge, and skills in pharmacy and specialty board certification of pharmacists. v This white paper adds focus and clarity to how BPS will approach the board certification of pharmacist specialists over the next five years. This timeline is critical as the goal will be to create a scalable foundation on which to build future board certification activities for pharmacists. -
![PHYSICIAN-SCIENTIST [MD/PHD] Who Better to Study Disease Than Those Who Know It Intimately? WHAT IS a PHYSICIAN SCIENTIST?](https://docslib.b-cdn.net/cover/7441/physician-scientist-md-phd-who-better-to-study-disease-than-those-who-know-it-intimately-what-is-a-physician-scientist-217441.webp)
PHYSICIAN-SCIENTIST [MD/PHD] Who Better to Study Disease Than Those Who Know It Intimately? WHAT IS a PHYSICIAN SCIENTIST?
PHYSICIAN-SCIENTIST [MD/PHD] Who better to study disease than those who know it intimately? WHAT IS A PHYSICIAN SCIENTIST? A physician scientist is a unique clinician who is part of a small cadre of physicians who usually work in academic health science centers and have a high impact on health care through discovery, translational and clinical research, and clinical practice. A physician scientist in Pathology and Laboratory Medicine is a laboratory physician who is trained in both scientific biomedical investigation and in pathology and/or laboratory medicine, often with clinical subspecialty training. On one hand, the physician scientist brings the rigors of scientific investigation into the patient care arena and on the other, the physician scientist’s contact with disease brings clinically relevant questions into the research arena to drive investigations into pathogenesis, prevention, diagnosis, prognosis, and treatment of disease. asip.org/CareerPath/ MEET ADEL MAHMOUD, MD, PHD P HYSICIAN . SCIENTIST. PROFESSOR . MENTOR . Princeton University Lecturer with rank of Professor in Molecular Biology and Public Policy, Woodrow Wilson School MD from the University of Cairo in 1963 PhD from the University of London, School of Hygiene and Tropical Medicine in 1971 Elected to membership of the American Society for Clinical Investigation in 1978, the Association of American Physicians in 1980 and the Institute of Medicine of the National Academy of Sciences in 1987. Received the Bailey K. Ashford Award of the A merican Society of Tropical Medicine and Hygiene in 1983, and the Squibb Award of the Infectious Diseases Society of America in 1984 . F e l l o w of the American College of Physicians and a member of the Expert Advisory Panel on Parasitic Diseases of the World Health Organization. -

Acquired Hemophilia A: Pathogenesis and Treatment
Bleeding disorders Acquired hemophilia A: pathogenesis and treatment P.W. Collins ABSTRACT Arthur Bloom Haemophilia Centre, Acquired hemophilia A is an autoimmune disease caused by an inhibitory antibody to factor VIII. The School of Medicine, severity of bleeding varies but patients remain at risk of life-threatening bleeding until the inhibitor Cardiff University, Heath Park, has been eradicated. The cornerstones of management are rapid and accurate diagnosis, control of Cardiff, UK bleeding, investigation for an underlying cause, and eradication of the inhibitor by immunosuppres - sion. Patients should be managed jointly with a specialist center even if they present without signifi - cant bleeding. Despite an extensive literature, few controlled data are available and management Hematology Education: guidelines are based on expert opinion. Recombinant factor VIIa and activated prothrombin complex the education program for the concentrate are equally efficacious for treating bleeds and both are superior to factor VIII or desmo - annual congress of the European pressin. Immunosuppression should be started as soon as the diagnosis is made. Commonly used reg - Hematology Association imens are steroids alone or combined with cytotoxic agents. Rituximab is being used more commonly but current evidence does not suggest that it improves outcomes or reduces side effects. 2012;6:65-72 Introduction Pathogenesis Acquired hemophilia A (AHA) is a bleed - AHA is associated with autoimmune dis - ing disorder caused by polyclonal IgG1 and eases, such as rheumatoid arthritis, polymyal - IgG4 autoantibodies to the factor VIII ( FVIII ) gia rheumatic, and systemic lupus erythe - A2 and C2 domain. Morbidity and mortality matosis; malignancy; pregnancy and dermato - are high secondary to age, underlying dis - logical disorders, such as pemphigoid. -

Bone Marrow (Stem Cell) Transplant for Sickle Cell Disease Bone Marrow (Stem Cell) Transplant
Bone Marrow (Stem Cell) Transplant for Sickle Cell Disease Bone Marrow (Stem Cell) Transplant for Sickle Cell Disease 1 Produced by St. Jude Children’s Research Hospital Departments of Hematology, Patient Education, and Biomedical Communications. Funds were provided by St. Jude Children’s Research Hospital, ALSAC, and a grant from the Plough Foundation. This document is not intended to take the place of the care and attention of your personal physician. Our goal is to promote active participation in your care and treatment by providing information and education. Questions about individual health concerns or specifi c treatment options should be discussed with your physician. For more general information on sickle cell disease, please visit our Web site at www.stjude.org/sicklecell. Copyright © 2009 St. Jude Children’s Research Hospital How did bone marrow (stem cell) transplants begin for children with sickle cell disease? Bone marrow (stem cell) transplants have been used for the treatment and cure of a variety of cancers, immune system diseases, and blood diseases for many years. Doctors in the United States and other countries have developed studies to treat children who have severe sickle cell disease with bone marrow (stem cell) transplants. How does a bone marrow (stem cell) transplant work? 2 In a person with sickle cell disease, the bone marrow produces red blood cells that contain hemoglobin S. This leads to the complications of sickle cell disease. • To prepare for a bone marrow (stem cell) transplant, strong medicines, called chemotherapy, are used to weaken or destroy the patient’s own bone marrow, stem cells, and infection fi ghting system.