Aldehyde Cyclocondensation Reaction in Natural Product Synthesis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Benzyl Ligand† Cite This: Chem
ChemComm View Article Online COMMUNICATION View Journal | View Issue Homoleptic organolanthanide compounds supported by the bis(dimethylsilyl)benzyl ligand† Cite this: Chem. Commun., 2017, 53,716 Kasuni C. Boteju, Arkady Ellern and Aaron D. Sadow* Received 21st November 2016, Accepted 12th December 2016 DOI: 10.1039/c6cc09304c www.rsc.org/chemcomm À A b-SiH functionalized benzyl anion [C(SiHMe2)2Ph] is obtained by A strategy for stabilizing coordinatively unsaturated rare earth deprotonation of HC(SiHMe2)2Ph with KCH2Ph or by reaction of amides has involved the incorporation of SiH groups, which 14 KOtBu and (Me2HSi)3CPh; LnI3(THF)n and three equivalents of this form labile secondary interactions with the lanthanide center. carbanion combine to provide homoleptic tris(alkyl)lanthanide Furthermore, the SiH moiety provides a powerful signature in 1 29 Creative Commons Attribution 3.0 Unported Licence. compounds Ln{C(SiHMe2)2Ph}3 (Ln = La, Ce, Pr, Nd) containing Hand Si NMR and IR spectra. This b-SiH strategy may also be secondary metal–ligand interactions. applied to alkyls, and the ligand C(SiHMe2)3 supports trivalent yttrium and divalent ytterbium and samarium homoleptic alkyls Synthesis of homoleptic organolanthanide complexes, particu- containing secondary Ln(H–Si interactions.15,16 Recently, we larly those of the early trivalent lanthanides (La–Nd), is challenging reported Ce{C(SiHMe2)3}3 as a precursor to a zwitterionic hydro- due to the large radii of these elements, polar bonding, high silylation catalyst.17 New chemistry might be accessed with alkyl charge, and high Lewis acidity.1 Such homoleptic compounds ligand variations that include both b-SiH and benzylic function- should be valuable for the synthesis of new catalysts and alities, and these groups could compete to enhance the homo- new materials,2 yet solvent- or donor-group-free, salt-free, and leptic compounds’ resistance to undesired ligand elimination This article is licensed under a thermally robust organolanthanide compounds are not readily pathways. -

Ganic Compounds
6-1 SECTION 6 NOMENCLATURE AND STRUCTURE OF ORGANIC COMPOUNDS Many organic compounds have common names which have arisen historically, or have been given to them when the compound has been isolated from a natural product or first synthesised. As there are so many organic compounds chemists have developed rules for naming a compound systematically, so that it structure can be deduced from its name. This section introduces this systematic nomenclature, and the ways the structure of organic compounds can be depicted more simply than by full Lewis structures. The language is based on Latin, Greek and German in addition to English, so a classical education is beneficial for chemists! Greek and Latin prefixes play an important role in nomenclature: Greek Latin ½ hemi semi 1 mono uni 1½ sesqui 2 di bi 3 tri ter 4 tetra quadri 5 penta quinque 6 hexa sexi 7 hepta septi 8 octa octo 9 ennea nona 10 deca deci Organic compounds: Compounds containing the element carbon [e.g. methane, butanol]. (CO, CO2 and carbonates are classified as inorganic.) See page 1-4. Special characteristics of many organic compounds are chains or rings of carbon atoms bonded together, which provides the basis for naming, and the presence of many carbon- hydrogen bonds. The valency of carbon in organic compounds is 4. Hydrocarbons: Compounds containing only the elements C and H. Straight chain hydrocarbons are named according to the number of carbon atoms: CH4, methane; C2H6 or H3C-CH3, ethane; C3H8 or H3C-CH2-CH3, propane; C4H10 or H3C-CH2- CH2-CH3, butane; C5H12 or CH3CH2CH2CH2CH3, pentane; C6H14 or CH3(CH2)4CH3, hexane; C7H16, heptane; C8H18, octane; C9H20, nonane; C10H22, CH3(CH2)8CH3, decane. -

Mann Et Al. Text 22
Catalytic Z-Selective Cross-Metathesis with Secondary Silyl- and Benzyl-Protected Allylic Ethers: Mechanistic Aspects and Applications to Natural Product Synthesis The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Mann, Tyler J., Alexander W. H. Speed, Richard R. Schrock, and Amir H. Hoveyda. “Catalytic Z-Selective Cross-Metathesis with Secondary Silyl- and Benzyl-Protected Allylic Ethers: Mechanistic Aspects and Applications to Natural Product Synthesis.” Angewandte Chemie International Edition 52, no. 32 (August 5, 2013): 8395-8400. As Published http://dx.doi.org/10.1002/anie.201302538 Publisher Wiley Blackwell Version Original manuscript Citable link http://hdl.handle.net/1721.1/84085 Terms of Use Creative Commons Attribution-Noncommercial-Share Alike 3.0 Detailed Terms http://creativecommons.org/licenses/by-nc-sa/3.0/ Catalytic Z-Selective Cross-Metathesis with Secondary Silyl- and Benzyl-Protected Allylic Ethers: Mechanistic Aspects and Applications to Natural Product Synthesis** Tyler J. Mann, Alexander W. H. Speed, Richard R. Schrock and Amir H. Hoveyda* [*] Prof. A. H. Hoveyda, T. J. Mann, Dr. A. W. H. Speed Department of Chemistry, Merkert Chemistry Center, Boston College, Chestnut Hill, MA 02467 (USA) Fax: (1) 617-552-1442 E-mail: [email protected] Prof. R. R. Schrock Department of Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02139 (USA) [**] Financial support was provided by the NIH (GM-59426). We are grateful to Robert V. O’Brien -

A General Protocol for the Formal Total Synthesis of (±)‐Strychnine A
Angewandte Communications Chemie International Edition:DOI:10.1002/anie.201611734 Annulations German Edition:DOI:10.1002/ange.201611734 Reaction of Donor-Acceptor Cyclobutanes with Indoles:AGeneral Protocol for the Formal Total Synthesis of ( )-Strychnine and the Total Synthesis of ( )-Akuammicine Æ Æ Liang-Wen Feng+,Hai Ren+,HuXiong,Pan Wang,Lijia Wang,* and Yong Tang* Abstract: Aligand-promoted catalytic [4+2] annulation reaction using indole derivatives and donor-acceptor (D-A) cyclobutanes is reported, thus providing an efficient and atom- economical access to versatile cyclohexa-fused indolines with excellent levels of diastereoselectivity and abroad substrate scope.Inthe presence of achiral SaBOXligand, excellent enantioselectivity was realized with up to 94%ee. This novel synthetic method is applied as ageneral protocol for the total synthesis of ( )-akuammicine and the formal total synthesis of Æ ( )-strychninefrom the same common-core scaffold. Æ Owing to their synthetic potential of accessing various cyclic compounds by formal [4 + n] cycloadditions,donor- acceptor (D-A) cyclobutanes have attracted increasing atten- Scheme 1. Anew route to access strychnos alkaloids. PG= protecting tion in recent years.[1,2] In 2013, Matsuo et al. reported an group, PMB =p-methoxybenzyl. elegant regioselective inter-and intramolecular formal [4+2] cycloaddition of cyclobutanones with indoles in 31–98% [a] yields with moderate to good diastereoselectivities.[2i] In 2016, Table 1: Reaction optimization. we demonstrated ahighly efficient formal -

Photoelectrocatalytic Selective Oxidation of 4-Methoxybenzyl Alcohol in Water By
Applied Catalysis B: Environmental 132–133 (2013) 535–542 Contents lists available at SciVerse ScienceDirect Applied Catalysis B: Environmental jo urnal homepage: www.elsevier.com/locate/apcatb Photoelectrocatalytic selective oxidation of 4-methoxybenzyl alcohol in water by TiO2 supported on titanium anodes a,∗ a b b c Levent Özcan , Sedat Yurdakal , Vincenzo Augugliaro , Vittorio Loddo , Simonetta Palmas , b b,∗∗ Giovanni Palmisano , Leonardo Palmisano a Kimya Bölümü, Fen-Edebiyat Fakültesi, Afyon Kocatepe Üniversitesi, Ahmet Necdet Sezer Kampüsü, 03200 Afyon, Turkey b “Schiavello-Grillone” Photocatalysis Group, Dipartimento di Energia, Ingegneria dell’Informazione, e Modelli Matematici (DEIM), University of Palermo, Viale delle Scienze, 90128 Palermo, Italy c Dipartimento di Ingegneria Meccanica, Chimica e dei Materiali, Università degli Studi di Cagliari, Via Marengo 2, 09123 Cagliari, Italy a r t i c l e i n f o a b s t r a c t Article history: The photoelectrocatalytic partial oxidation of 4-methoxybenzyl alcohol in aqueous solution irradiated Received 31 October 2012 by near-UV light was carried out in a three-electrode batch reactor. TiO2 films were either deposited by Received in revised form dip-coating of a TiO2 sol onto a Ti foil and subsequent calcination or generated on Ti plates by ther- 14 December 2012 ◦ mal oxidation in air at 400–700 C. The effects of the anode preparation method and bias potential Accepted 18 December 2012 values on conversion and selectivity to the corresponding aldehyde were investigated. The photo- Available online 27 December 2012 electrocatalytic results were compared with the photocatalytic and electrocatalytic ones. The results indicated that no reaction occurred during the electrocatalytic experiments, whereas the photocatalytic Keywords: Photoelectrocatalysis reactivity was positively influenced by the application of a small bias (0.75 V vs. -

(). X (O) 58 Field of Search
United States Patent (19) 11) 4,197,307 Gallay et al. 45) Apr. 8, 1980 (54) 2-ALKYLTHIO-, 2-ALKYLSULPHINYL AND 2-ALKYLSULFONYL-6-PHENYELBEN R2 ZIMEDAZOLES AS ANTHELMINTEC AGENTS R3 N (Y), )-i-R 75 Inventors: Jean-Jacques Gallay, Magden; X (O) Manfred Kthne, Pfeffingen; Alfred R4 R Meyer, Basel; Oswald Rechsteiner, Binningen; Max Schellenbaum, in which Muttenz, all of Switzerland R is an alkyl group having 1 to 6 carbon atoms, an alkenyl group having 3 to 5 carbon atoms, an alkynyl group having 3 to 5 carbon atoms or a benzyl group, 73) Assignee: Ciba-Geigy Corporation, Ardsley, which is unsubstituted or monosubstituted to disubsti tuted by a methyl group, halogen or a nitro group; R1 is hydrogen, an alkanoyl group having 1 to 4 carbon atoms, an alkoxycarbonyl group having 1 to 4 carbon (21) Appl. No.: 894,973 atoms, a N,N-dialkylcarbamoyl group or N,N-dialk ylthiocarbamoyl group, each having 1 to 4 carbon atoms in the alkyl groups, an alkylsulphonyl group 22 Filed: Apr. 10, 1978 having 1 to 4 carbon atoms, a benzoyl group, a phe nylsulphonyl group, a 4-methylphenylsulphony 30 Foreign Application Priority Data group or the radical, Apr. 12, 1977 LU) Luxembourg ........................... 77120 Mar. 15, 1978 LU Luxembourg ........................... 79232 R3 N (Y), >--R 51 Int. C.? ................... A61K 31/415; C07D 235/28 52 U.S. C. ................................ 424/273 B; 548/305; 548/328; 548/329 (). X (O) 58 Field of Search ............................... 548/329, 328; 424/273 R, 273 B in which Q is a carbonyl group, a thiocarbonyl group or an 56) References Cited oxalyl group; U.S. -
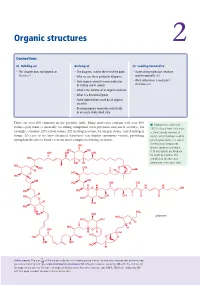
Organic Structures 2
Organic structures 2 Connections Building on Arriving at Looking forward to • This chapter does not depend on • The diagrams used in the rest of the book • Ascertaining molecular structure Chapter 1 • Why we use these particular diagrams spectroscopically ch3 • How organic chemists name molecules • What determines a molecule’s in writing and in speech structure ch4 • What is the skeleton of an organic molecule • What is a functional group • Some abbreviations used by all organic chemists • Drawing organic molecules realistically in an easily understood style There are over 100 elements in the periodic table. Many molecules contain well over 100 ■ Palytoxin was isolated in atoms—palytoxin (a naturally occurring compound with potential anticancer activity), for 1971 in Hawaii from Limu make example, contains 129 carbon atoms, 221 hydrogen atoms, 54 oxygen atoms, and 3 nitrogen o Hane (‘deadly seaweed of atoms. It’s easy to see how chemical structures can display enormous variety, providing Hana’), which had been used to enough molecules to build even the most complicated living creatures. poison spear points. It is one of the most toxic compounds OH known, requiring only about HO OH 0.15 micrograms per kilogram OH OH OH for death by injection. The HO complicated structure was O OH H determined a few years later. HO OH OH O H H O O HO HO H OH OH HO OH OH H H H H O N N OH HO HO H HO OH OH O O O HO OH H OH HO OH NH2 HO OH HO palytoxin HO OH O H HO H OH O H H H HO HO OH O O OH OH HO H Online support. -

Studies Toward the Total Synthesis of (+)-Pyrenolide D Honors Research
Studies toward the Total Synthesis of (+)-Pyrenolide D Honors Research Thesis Presented in partial fulfillment of the requirements for graduation with honors research distinction in Chemistry in the undergraduate colleges of The Ohio State University by Adam Doane The Ohio State University 24 May 2012 Project Advisor: Dr. Christopher Callam, Department of Chemistry i Abstract Spiro lactones have had an extensive impact in both medicinal and organismal biology. Since their discovery, various methods for preparing spiro lactones have been studied and investigated. One of these spiro lactones, pyrenolide D, which is one of the four metabolites, A- D, was isolated from Pyrenophora teres by Nukina and Hirota in 1992 and shows cytotoxicity towards HL-60 cells at IC50 4 µg/ mL. The structural determination, via spectroscopic methods, reported that unlike pyrenolides A-C, pyrenolide D contained a rare, highly oxygenated tricyclic spiro-γ-lactone structure and did not contain the macrocyclic lactone seen in the other pyrenolides. The purpose of this project is the determination of a cost effective and efficient synthesis of pyrenolide D from the naturally occurring sugar, D-xylose. Herein we report studies towards the total synthesis of pyrenolide D. i Acknowledgments I would like to thank my research advisor, Dr. Christopher Callam for his constant help, encouragement, and inspiration throughout the past 3 years. He has sacrificed an immeasurable number of hours answering questions and providing support for the duration of this project. I would also like to thank The Ohio State University Department of Chemistry for financial support as well as Chemical Abstracts Service, the William Marshall MacNevin Memorial foundation, and Mr. -

12: Conjugated and Aromatic Molecules
(3/94)(9,10/96)(3,4,5/04) Neuman Chapter 12 Chapter 12 Conjugated and Aromatic Molecules from Organic Chemistry by Robert C. Neuman, Jr. Professor of Chemistry, emeritus University of California, Riverside [email protected] <http://web.chem.ucsb.edu/~neuman/orgchembyneuman/> Chapter Outline of the Book ************************************************************************************** I. Foundations 1. Organic Molecules and Chemical Bonding 2. Alkanes and Cycloalkanes 3. Haloalkanes, Alcohols, Ethers, and Amines 4. Stereochemistry 5. Organic Spectrometry II. Reactions, Mechanisms, Multiple Bonds 6. Organic Reactions *(Not yet Posted) 7. Reactions of Haloalkanes, Alcohols, and Amines. Nucleophilic Substitution 8. Alkenes and Alkynes 9. Formation of Alkenes and Alkynes. Elimination Reactions 10. Alkenes and Alkynes. Addition Reactions 11. Free Radical Addition and Substitution Reactions III. Conjugation, Electronic Effects, Carbonyl Groups 12. Conjugated and Aromatic Molecules 13. Carbonyl Compounds. Ketones, Aldehydes, and Carboxylic Acids 14. Substituent Effects 15. Carbonyl Compounds. Esters, Amides, and Related Molecules IV. Carbonyl and Pericyclic Reactions and Mechanisms 16. Carbonyl Compounds. Addition and Substitution Reactions 17. Oxidation and Reduction Reactions 18. Reactions of Enolate Ions and Enols 19. Cyclization and Pericyclic Reactions *(Not yet Posted) V. Bioorganic Compounds 20. Carbohydrates 21. Lipids 22. Peptides, Proteins, and α−Amino Acids 23. Nucleic Acids ************************************************************************************** *Note: Chapters marked with an (*) are not yet posted. 0 (3/94)(9,10/96)(3,4,5/04) Neuman Chapter 12 12: Conjugated and Aromatic Molecules 12.1 Conjugated Molecules 12-4 1,3-Butadiene (12.1A) 12-4 Atomic Orbital Overlap in 1,3-Butadiene Molecular Orbitals The Bonding M.O.'s Conformations Other Alternating Multiple Bonds Pentadienes (12.1B) 12-7 1,3-Pentadiene 1,4-Pentadiene 1,2-Pentadiene. -

Nomenclature of Substituted Benzene Rings
Chem 263 Oct. 5, 2010 Nomenclature of substituted benzene rings When you have two substituents on a benzene ring, ortho, meta, and para are used to tell where the second substitution is relative to the first one. X ortho ortho meta meta para Ortho refers to 1,2-substitution and is abbreviated o- Meta refers to 1,3-substitution and is abbreviated m- Para refers to 1,4-substitution and is abbreviated p- Examples: O OH OH C p-aminophenol (more correct, OH has priority) or p-methoxybenzoic acid p-hydroxyaniline (this is an ether group, NH2 OCH3 specifically methoxy) p-Aminophenol: The amine and hydroxyl group are in the 1 and 4 positions, so they are para to each other. The parent structure in this molecule can be either aniline or phenol. For this course, it doesn’t matter which parent structure you pick in the nomenclature with these substituents. Usually when naming the substituents, the atomic number takes priority, but there are many exceptions for historical reasons. Name: m-bromophenol or 3-bromophenol In this compound, the –OH (hydroxy) and –Br are in the 1 and 3 positions, so they are meta (or abbreviated m) to each other. The parent structure is phenol (phenol is a benzene with a hydroxyl group directly attached, not to be confused with phenyl which means just a benzene ring as a substituent), so we call this meta-bromophenol. O OH O OH C C OH O O 2-hydroxybenzoic acid aspirin aka salicylic acid acetylsalicylic acid O H OCH3 OH Nomenclature: 4-hydroxy-3-methoxybenzaldehyde (or vanillin from vanilla extract). -

17.8013.01000 Sixty-Fifth Legislative Assembly SENATE BILL NO
17.8013.01000 Sixty-fifth Legislative Assembly SENATE BILL NO. 2096 of North Dakota Introduced by Judiciary Committee (At the request of the State Board of Pharmacy) 1 A BILL for an Act to amend and reenact sections 19-03.1-05, 19-03.1-07, 19-03.1-11, and 2 19-03.1-13 of the North Dakota Century Code, relating to the scheduling of controlled 3 substances; and to declare an emergency. 4 BE IT ENACTED BY THE LEGISLATIVE ASSEMBLY OF NORTH DAKOTA: 5 SECTION 1. AMENDMENT. Section 19-03.1-05 of the North Dakota Century Code is 6 amended and reenacted as follows: 7 19-03.1-05. Schedule I. 8 1. The controlled substances listed in this section are included in schedule I. 9 2. Schedule I consists of the drugs and other substances, by whatever official name, 10 common or usual name, chemical name, or brand name designated, listed in this 11 section. 12 3. Opiates. Unless specifically excepted or unless listed in another schedule, any of the 13 following opiates, including their isomers, esters, ethers, salts, and salts of isomers, 14 esters, and ethers, whenever the existence of those isomers, esters, ethers, and salts 15 is possible within the specific chemical designation: 16 a. Acetyl-alpha-methylfentanyl (also known as N-[1-(1-methyl-2-phenethyl)-4- 17 piperidinyl]-N-phenylacetamide). 18 b. Acetylfentanyl (also known as N-(1-phenethylpiperidin-4-yl)-N-phenylacetamide). 19 c. Acetylmethadol. 20 d.b. Allylprodine. 21 e.c. Alphacetylmethadol. 22 f.d. Alphameprodine. 23 g.e. -

1 Chapter 11: Arenes and Aromaticity 11.1: Benzene
Chapter 11: Arenes and Aromaticity 11.1: Benzene - C6H6 H H H H H H H H H H H H 11.2: Kekulé and the Structure of Benzene Kekule benzene: two forms are in rapid equilibrium 154 pm 134 pm • All bonds are 140 pm (intermediate between C-C and C=C) • C–C–C bond angles are 120° • Structure is planar, hexagonal 245 11.3: A Resonance Picture of Bonding in Benzene resonance hybrid 6 π-electron delocalized over 6 carbon atoms 11.4: The Stability of Benzene Aromaticity: cyclic conjugated organic compounds such as benzene, exhibit special stability due to resonance delocalization of π -electrons. 246 1 Heats of hydrogenation (Fig. 11.2, p. 425) + H2 + 120 KJ/mol + 2 H2 + 230 KJ/mol calc'd value= 240 KJ/mol 10 KJ/mol added stability + 3 H 2 + 208 KJ/mol calc'd value= 360 KJ/mol 152 KJ/mol added stability + 3 H2 + 337 KJ/mol 1,3,5-Hexatriene - conjugated but not cyclic Resonance energy of benzene is 129 - 152 KJ/mol 247 11.5: An Orbital Hybridization View of Bonding in Benzene • Benzene is a planar, hexagonal cyclic hydrocarbon • The C–C–C bond angles are 120° = sp2 hybridized • Each carbon possesses an unhybridized p-orbital, which makes up the conjugated π-system. • The six π-electrons are delocalized through the π-system 11.6: The π Molecular Orbitals of Benzene - the aromatic system of benzene consists of six p-orbitals (atomic orbitals). Benzene must have six molecular orbitals. 248 2 !6 !4 !5 six p-orbitals !2 !3 !1 Degenerate orbitals: : zero nodes orbitals that have the Ψ1 Bonding and : one node same energy Ψ2 Ψ3 and : two nodes Ψ4 Ψ5 Anti-bonding