Gall-ID: Tools for Genotyping Gall-Causing Phytopathogenic Bacteria
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Revisiting the Taxonomy of Allorhizobium Vitis (Ie
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.19.423612; this version posted December 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Revisiting the taxonomy of Allorhizobium vitis (i.e. Agrobacterium vitis) using genomics - emended description of All. vitis sensu stricto and description of Allorhizobium ampelinum sp. nov. Nemanja Kuzmanović1,*, Enrico Biondi2, Jörg Overmann3, Joanna Puławska4, Susanne Verbarg3, Kornelia Smalla1, Florent Lassalle5,6,* 1Julius Kühn-Institut, Federal Research Centre for Cultivated Plants (JKI), Institute for Epidemiology and Pathogen Diagnostics, Messeweg 11-12, 38104 Braunschweig, Germany 2Alma Mater Studiorum - University of Bologna, Viale G. Fanin, 42, 40127 Bologna, Italy 3Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures, Inhoffenstrasse 7B, 38124 Braunschweig, Germany 4Research Institute of Horticulture, ul. Konstytucji 3 Maja 1/3, 96-100 Skierniewice, Poland 5Imperial College London, St-Mary’s Hospital campus, Department of Infectious Disease Epidemiology, Praed Street, London W2 1NY, UK; Imperial College London, St-Mary’s Hospital campus, MRC Centre for Global Infectious Disease Analysis, Praed Street, London W2 1NY, United Kingdom 6Wellcome Sanger Institute, Pathogens and Microbes Programme, Wellcome Genome Campus, Hinxton, Saffron Walden, CB10 1RQ, United Kingdom *Corresponding authors. Contact: [email protected], [email protected] (N. Kuzmanovid); [email protected] (F. Lassalle) bioRxiv preprint doi: https://doi.org/10.1101/2020.12.19.423612; this version posted December 21, 2020. -

Biocontrol of Crown Gall by Rhizobium Rhizogenes
agronomy Case ReportReport Biocontrol of Crown Gall byby RhizobiumRhizobium rhizogenesrhizogenes:: Challenges in Biopesticide Commercialisation 1 2, Allen Kerr 1 and Gary Bullard 2,** 1 Department of Plant Pathology, University of Adelaide, Adelaide, SA 5064, Australia; [email protected] 1 Department of Plant Pathology, University of Adelaide, Adelaide SA 5064, Australia; [email protected] 2 Bio-Care Technology Pty Ltd., Myocum, NSW 2481, Australia 2 Bio-Care Technology Pty Ltd., Myocum NSW 2481, Australia * Correspondence: [email protected] * Correspondence: [email protected] Received: 18 June 2020 2020;; Accepted: 30 July 2020 2020;; Published: 3 3 August August 2020 2020 Abstract: The biocontrolbiocontrol of crown gall has been practisedpractised in AustraliaAustralia for 4848 years.years. Control is so eefficientfficient thatthat itit is is di difficultfficult to to find find a galleda galled stone stone fruit fruit tree, tree, when when previously, previously, crown crown gall had gall been had abeen major a problem.major problem. This paper This explainspaper explains how it works how andit works why onlyand pathogenswhy only arepathog inhibited.ens are A inhibited. commercial A biopesticidecommercial biopesticide is available inis Australia,available in Canada, Australia, Chile, Canada, New Zealand,Chile, New Turkey, Zealand, the USA, Turkey, South the Africa USA, andSouth Japan. Africa The and challenges Japan. The of commercialisingchallenges of commercialising a biopesticide area biopesticide outlined. Rigid -

Revised Taxonomy of the Family Rhizobiaceae, and Phylogeny of Mesorhizobia Nodulating Glycyrrhiza Spp
Division of Microbiology and Biotechnology Department of Food and Environmental Sciences University of Helsinki Finland Revised taxonomy of the family Rhizobiaceae, and phylogeny of mesorhizobia nodulating Glycyrrhiza spp. Seyed Abdollah Mousavi Academic Dissertation To be presented, with the permission of the Faculty of Agriculture and Forestry of the University of Helsinki, for public examination in lecture hall 3, Viikki building B, Latokartanonkaari 7, on the 20th of May 2016, at 12 o’clock noon. Helsinki 2016 Supervisor: Professor Kristina Lindström Department of Environmental Sciences University of Helsinki, Finland Pre-examiners: Professor Jaakko Hyvönen Department of Biosciences University of Helsinki, Finland Associate Professor Chang Fu Tian State Key Laboratory of Agrobiotechnology College of Biological Sciences China Agricultural University, China Opponent: Professor J. Peter W. Young Department of Biology University of York, England Cover photo by Kristina Lindström Dissertationes Schola Doctoralis Scientiae Circumiectalis, Alimentariae, Biologicae ISSN 2342-5423 (print) ISSN 2342-5431 (online) ISBN 978-951-51-2111-0 (paperback) ISBN 978-951-51-2112-7 (PDF) Electronic version available at http://ethesis.helsinki.fi/ Unigrafia Helsinki 2016 2 ABSTRACT Studies of the taxonomy of bacteria were initiated in the last quarter of the 19th century when bacteria were classified in six genera placed in four tribes based on their morphological appearance. Since then the taxonomy of bacteria has been revolutionized several times. At present, 30 phyla belong to the domain “Bacteria”, which includes over 9600 species. Unlike many eukaryotes, bacteria lack complex morphological characters and practically phylogenetically informative fossils. It is partly due to these reasons that bacterial taxonomy is complicated. -

Octopine Synthase Mrna Isolated from Sunflower Crown Gall Callus Is
Proc. Nati Acad. Sci. USA Vol. 79, pp. 86-90, January 1982 Biochemistry Octopine synthase mRNA isolated from sunflower crown gall callus is homologous to the Ti plasmid of Agrobacterium tumefaciens (recombinant plasmid/hybridization/mRNA size/in vitro translation/immunoprecipitation) NORIMOTO MURAI AND JOHN D. KEMP Department of Plant Pathology, University ofWisconsin, and Plant Disease Research Unit, ARS, USDA, Madison, Wisconsin 53706 Communicated by Folke Skoog, September 14, 1981 ABSTRACT We have shown that the structural gene for oc- families have been identified. The enzymes responsible for the topine synthase (a crown gall-specific enzyme) is located in a cen- synthesis of the first two have been purified and characterized tral portion ofthe T-DNA that came from the Ti plasmid ofAgro- (17, 18). bacterium tumefaciens and is expressed after it has been trans- The particular opine family present in a crown gall cell cor- ferred to the plant cells. Polyadenylylated RNA was prepared relates with a particular Ti plasmid rather than with the host from polysomes isolated from an octopine-producing crown gall plant (19, 20). Analysis ofdeletion mutants of an octopine-type callus and purified by selective hybridization to one offive recom- Ti plasmid has shown that a gene controlling octopine produc- binant plasmids. Each such plasmid contained a different frag- tion is located on a 14.6-kbp fragment ofthe T-DNA generated ment ofT-DNA ofpTi-15955 (octopine-type Ti plasmid). Purified by Sin 1 (21) (Fig. 1). Detailed examination ofthe physical or- mRNA was translated in vitro in rabbit reticulocyte lysates, and ganization of T-DNA in octopine-type tumor lines has shown the translation products were immunoprecipitated with antibody that octopine production is correlated with the presence ofthe against octopine synthase. -

(12) United States Patent (10) Patent No.: US 7,112,717 B2 Valentin Et Al
US007 112717B2 (12) United States Patent (10) Patent No.: US 7,112,717 B2 Valentin et al. (45) Date of Patent: Sep. 26, 2006 (54) HOMOGENTISATE PRENYLTRANSFERASE 2004/0045051 A1 3/2004 Norris et al. GENE (HPT2). FROM ARABIDOPSIS AND USES THEREOF FOREIGN PATENT DOCUMENTS (75) Inventors: Henry E. Valentin, Chesterfield, MO CA 2339519 2, 2000 (US); Tyamagondlu V. Venkatesh, St. CA 2343919 3, 2000 Louis, MO (US); Karunanandaa CA 237,2332 11 2000 Balasulojini, Creve Coeur, MO (US) DE 19835 219 A1 8, 1998 (73) Assignee: Monsanto Technology LLC, St. Louis, EP O 531 639 A2 3, 1993 MO (US) E. 8. G. O. (*) Notice: Subject to any disclaimer, the term of this EP O 723 O17 A2 T 1996 patent is extended or adjusted under 35 EP O 763 542 A2 3, 1997 U.S.C. 154(b) by 404 days. EP 1 033. 405 A2 9, 2000 EP 1 063. 297 A1 12/2000 (21) Appl. No.: 10/391,363 FR 2 778 527 11, 1999 GB 560.529 4f1944 (22) Filed: Mar. 18, 2003 WO WO 91/02059 2, 1991 WO WO 91,09128 6, 1991 (65) Prior Publication Data WO WO 91/13078 9, 1991 WO WO 93/18158 9, 1993 US 2003/0213017 A1 Nov. 13, 2003 WO WO 94,11516 5, 1994 O O WO WO 94/12014 6, 1994 Related U.S. Application Data WO WO94, 18337 8, 1994 (60) Provisional application No. 60/365,202, filed on Mar. WO WO95/08914 4f1995 19, 2002. WO WO95/18220 7, 1995 WO WO95/23863 9, 1995 (51) Int. -
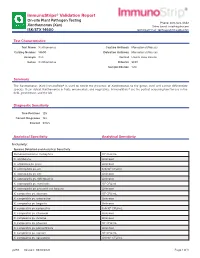
Immunostrips® Validation Report
ImmunoStrips® Validation Report On-site Plant Pathogen Testing Phone: 800-622-4342 Xanthomonas (Xan) Sales Email: [email protected] ISK/STX 14600 Technical Email: [email protected] Test Characteristics Test Name Xanthomonas Capture Antibody Monoclonal (Mouse) Catalog Number 14600 Detection Antibody Monoclonal (Mouse) Acronym Xan Format Lateral Flow Device Genus Xanthomonas Diluents SEB1 Sample Dilution 1:20 Summary The Xanthomonas (Xan) ImmunoStrip® is used to detect the presence of Xanthomonas to the genus level and cannot differentiate species. It can detect Xanthomonas in fruits, ornamentals, and vegetables. ImmunoStrips® are the perfect screening tool for use in the field, greenhouse, and the lab. Diagnostic Sensitivity True Positives 125 Correct Diagnoses 122 Percent 97.6% Analytical Specificity Analytical Sensitivity Inclusivity: Species Detected and Analytical Sensitivity Stenotrophomonas maltophilia 106 CFU/mL X. albilibeans Unknown X. arboricola pv. pruni Unknown X. axonopodis pv. alii 6.9x104 CFU/mL X. axonopodis pv. citri Unknown X. axonopodis pv. diffenbachia Unknown X. axonopodis pv. manihotis 106 CFU/mL X. axonopodis pv. phaseoli var. fuscans Unknown X. campestris pv. aberrans 105 CFU/mL X. campestris pv. armoraciae Unknown X. campestris pv. begonia Unknown X. campestris pv. campestris 6.4x104 CFU/mL X. campestris pv. citromelo Unknown X. campestris pv. incanae Unknown X. campestris pv. phaseoli 105 CFU/mL X. campestris pv. poinsettiicola Unknown X. campestris pv. raphani 105 CFU/mL X. campestris pv. vesicatoria 4.0x105 CFU/mL p293 Revised: 08/18/2021 Page 1 of 3 Species Detected and Analytical Sensitivity X. campestris pv. vitians 105 CFU/mL X. campestris pv. zinnia 107 CFU/mL X. cannabis 1.6x104 CFU/mL X. -

Plant Root 11:33-39
www.plantroot.org 33 Original research article Biolistic-mediated plasmid-free transformation for induction of hairy roots Revi einw a tobaccorticle plantsShort report Gulnar Yasybaeva, Zilya Vershinina, Bulat Kuluev, Elena Mikhaylova, Andrey Baymiev and Aleksey Chemeris Institute of Biochemistry and Genetics, Ufa Scientific Centre, Russian Academy of Sciences (IBG USC RAS), pr. Oktyabrya 71, 450054, Ufa, Russia Corresponding author. G. Yasybaeva, E-mail: [email protected], Phone: +7-937-480-9956 Received on December 13, 2016; Accepted on March 8, 2017 Abstract: T-DNA of Ri-plasmid from Agrobacteri- transformed plant tissue, specifically, promotes the um rhizogenes is able to trigger the hairy root syn- appearance of hairy roots. Hairy roots culture (HRC) drome in infected plants. This natural phenomenon can be used for production of various secondary is used to generate hairy root cultures predomi- metabolites such as alkaloids, terpenoids, phenols, nantly only in dicotyledonous plants. We propose a glycosides (Sharifi et al. 2014). Some of them are a new method of hairy roots induction without Agro- source of pharmaceutical raw materials or other bacterium-mediated transformation. The 5461 bp economically valuable compounds. HRCs also can T-DNA region from A. rhizogenes A4 strain with all be used as recombinant proteins producers (Ono and four rol genes was amplified using primers contain- Tian 2011). ing sequences for left and right T-DNA borders on In natural conditions soil bacteria A. rhizogenes their 3’-ends. This amplicon was used for direct infects healthy plants by penetrating through injured transformation of tobacco leaf discs without A. rhi- roots. Thereby simple way of making hairy roots is zogenes and binary vectors. -

2010.-Hungria-MLI.Pdf
Mohammad Saghir Khan l Almas Zaidi Javed Musarrat Editors Microbes for Legume Improvement SpringerWienNewYork Editors Dr. Mohammad Saghir Khan Dr. Almas Zaidi Aligarh Muslim University Aligarh Muslim University Fac. Agricultural Sciences Fac. Agricultural Sciences Dept. Agricultural Microbiology Dept. Agricultural Microbiology 202002 Aligarh 202002 Aligarh India India [email protected] [email protected] Prof. Dr. Javed Musarrat Aligarh Muslim University Fac. Agricultural Sciences Dept. Agricultural Microbiology 202002 Aligarh India [email protected] This work is subject to copyright. All rights are reserved, whether the whole or part of the material is concerned, specifically those of translation, reprinting, re-use of illustrations, broadcasting, reproduction by photocopying machines or similar means, and storage in data banks. Product Liability: The publisher can give no guarantee for all the information contained in this book. The use of registered names, trademarks, etc. in this publication does not imply, even in the absence of a specific statement, that such names are exempt from the relevant protective laws and regulations and therefore free for general use. # 2010 Springer-Verlag/Wien Printed in Germany SpringerWienNewYork is a part of Springer Science+Business Media springer.at Typesetting: SPI, Pondicherry, India Printed on acid-free and chlorine-free bleached paper SPIN: 12711161 With 23 (partly coloured) Figures Library of Congress Control Number: 2010931546 ISBN 978-3-211-99752-9 e-ISBN 978-3-211-99753-6 DOI 10.1007/978-3-211-99753-6 SpringerWienNewYork Preface The farmer folks around the world are facing acute problems in providing plants with required nutrients due to inadequate supply of raw materials, poor storage quality, indiscriminate uses and unaffordable hike in the costs of synthetic chemical fertilizers. -

By USDA APHIS BRS Document Control Officer at 2:18 Pm, Dec 11
CBI Deleted EXECUTIVE SUMMARY The Animal and Plant Health Inspection Service (APHIS) of the United States (U.S.) Department of Agriculture (USDA) has responsibility under the Plant Protection Act (Title IV Pub. L. 106-224, 114 Stat. 438, 7 U.S.C. § 7701-7772) to prevent the introduction and dissemination of plant pests into the U.S. APHIS regulation 7 CFR § 340.6 provides that an applicant may petition APHIS to evaluate submitted data to determine that a particular regulated article does not present a plant pest risk and no longer should be regulated. If APHIS determines that the regulated article does not present a plant pest risk, the petition is granted, thereby allowing unrestricted introduction of the article. Monsanto Company is submitting this request to APHIS for a determination of nonregulated status for the new biotechnology-derived maize product, MON 87429, any progeny derived from crosses between MON 87429 and conventional maize, and any progeny derived from crosses of MON 87429 with biotechnology-derived maize that have previously been granted nonregulated status under 7 CFR Part 340. Product Description Monsanto Company has developed herbicide tolerant MON 87429 maize, which is tolerant to the herbicides dicamba, glufosinate, aryloxyphenoxypropionate (AOPP) acetyl coenzyme A carboxylase (ACCase) inhibitors (so called “FOPs” herbicides such as quizalofop) and 2,4-dichlorophenoxyacetic acid (2,4-D). In addition, it provides tissue-specific glyphosate tolerance to facilitate the production of hybrid maize seeds. MON 87429 contains -

Monsanto Company Insect Resistant Soybean
RELEASE OF INFORMATION Monsanto is submitting the information in this petition for review by the USDA as part of the regulatory process. By submitting this information, Monsanto does not authorize its release to any third party. In the event the USDA receives a Freedom of Information Act request, pursuant to 5 U.S.C. § 552, and 7 CFR Part 1, covering all or some of this information, Monsanto expects that, in advance of the release of the document(s), USDA will provide Monsanto with a copy of the material proposed to be released and the opportunity to object to the release of any information based on appropriate legal grounds, e.g. responsiveness, confidentiality, and/or competitive concerns. Monsanto expects that no information that has been identified as CBI (confidential business information), will be provided to any third party. Monsanto understands that a CBI- deleted copy of this information may be made available to the public in a reading room and by individual request, as part of a public comment period. Except in accordance with the foregoing, Monsanto does not authorize the release, publication or other distribution of this information (including website posting) without Monsanto's prior notice and consent. 2009 Monsanto Company. All Rights Reserved. This document is protected under copyright law. This document is for use only by the regulatory authority to which this has been submitted by Monsanto Company, and only in support of actions requested by Monsanto Company. Any other use of this material, without prior written consent of Monsanto, is strictly prohibited. By submitting this document, Monsanto does not grant any party or entity any right to license or to use the information or intellectual property described in this document. -

Introduction to Plant Pathology {Richard N. Strange
Introduction to Plant Pathology Introduction to Plant Pathology Richard N. Strange University College London Copyright ß 2003 by John Wiley & Sons Ltd, The Atrium, Southern Gate, Chichester, West Sussex PO19 8SQ, England National l01243 779777 International (þ44) 1243 779777 e-mail (for orders and customer service enquiries): [email protected] Visit our Home Page on http://www.wiley.co.uk or http://www.wiley.com All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording, scanning or otherwise, except under the terms of the Copyright, Designs and Patents Act 1988 or under the terms of a licence issued by the Copyright Licensing Agency, 90 Tottenham Court Road, London, UK W1P 9 HE, without the permission in writing of the publisher. Other Wiley Editorial Offices John Wiley & Sons, Inc., 605 Third Avenue, New York, NY 10158-0012, USA Wiley-VCH Verlag GmbH, Pappelallee 3, D-69469 Weinheim, Germany John Wiley & Sons (Australia) Ltd, 33 Park Road, Milton, Queensland 4064, Australia John Wiley & Sons (Asia) Pte Ltd, 2 Clementi Loop #02-01, Jin Xing Distripark, Singapore 0512 John Wiley & Sons (Canada) Ltd, 22 Worcester Road, Rexdale, Ontario M9W 1L1, Canada Wiley also publishes its books in a variety of electronic formats. Some content that appears in print may not be available in electronic books. Library of Congress Cataloging-in-Publication Data Strange, Richard N. Introduction to plant pathology / Richard N. Strange. p. cm. Includes bibliographical references and index. ISBN 0-470-84972-X (Cloth : alk. -

A Review of Root Mat and Crown Gall Diseases to Inform Research on Control of Tomato Root Mat
Protected tomato – A review of root mat and crown gall diseases to inform research on control of tomato root mat Tim O’Neill and Sarah Mayne, ADAS John Elphinstone, Fera May 2016 PE 029 Contents Background ........................................................................................................................... 1 Summary .............................................................................................................................. 2 Technical review ................................................................................................................... 5 1. Occurrence of root mat ........................................................................................... 5 a) Rosaceous crops ................................................................................................. 6 b) Cucumber ............................................................................................................ 8 c) Tomato .............................................................................................................. 10 d) Other protected edible crops ............................................................................. 12 2. Occurrence of crown gall ...................................................................................... 12 a) Rosaceous crop ................................................................................................ 13 b) Other plant families ........................................................................................... 14 3.