View Preprint
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Revisiting the Taxonomy of Allorhizobium Vitis (Ie
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.19.423612; this version posted December 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Revisiting the taxonomy of Allorhizobium vitis (i.e. Agrobacterium vitis) using genomics - emended description of All. vitis sensu stricto and description of Allorhizobium ampelinum sp. nov. Nemanja Kuzmanović1,*, Enrico Biondi2, Jörg Overmann3, Joanna Puławska4, Susanne Verbarg3, Kornelia Smalla1, Florent Lassalle5,6,* 1Julius Kühn-Institut, Federal Research Centre for Cultivated Plants (JKI), Institute for Epidemiology and Pathogen Diagnostics, Messeweg 11-12, 38104 Braunschweig, Germany 2Alma Mater Studiorum - University of Bologna, Viale G. Fanin, 42, 40127 Bologna, Italy 3Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures, Inhoffenstrasse 7B, 38124 Braunschweig, Germany 4Research Institute of Horticulture, ul. Konstytucji 3 Maja 1/3, 96-100 Skierniewice, Poland 5Imperial College London, St-Mary’s Hospital campus, Department of Infectious Disease Epidemiology, Praed Street, London W2 1NY, UK; Imperial College London, St-Mary’s Hospital campus, MRC Centre for Global Infectious Disease Analysis, Praed Street, London W2 1NY, United Kingdom 6Wellcome Sanger Institute, Pathogens and Microbes Programme, Wellcome Genome Campus, Hinxton, Saffron Walden, CB10 1RQ, United Kingdom *Corresponding authors. Contact: [email protected], [email protected] (N. Kuzmanovid); [email protected] (F. Lassalle) bioRxiv preprint doi: https://doi.org/10.1101/2020.12.19.423612; this version posted December 21, 2020. -

Biocontrol of Crown Gall by Rhizobium Rhizogenes
agronomy Case ReportReport Biocontrol of Crown Gall byby RhizobiumRhizobium rhizogenesrhizogenes:: Challenges in Biopesticide Commercialisation 1 2, Allen Kerr 1 and Gary Bullard 2,** 1 Department of Plant Pathology, University of Adelaide, Adelaide, SA 5064, Australia; [email protected] 1 Department of Plant Pathology, University of Adelaide, Adelaide SA 5064, Australia; [email protected] 2 Bio-Care Technology Pty Ltd., Myocum, NSW 2481, Australia 2 Bio-Care Technology Pty Ltd., Myocum NSW 2481, Australia * Correspondence: [email protected] * Correspondence: [email protected] Received: 18 June 2020 2020;; Accepted: 30 July 2020 2020;; Published: 3 3 August August 2020 2020 Abstract: The biocontrolbiocontrol of crown gall has been practisedpractised in AustraliaAustralia for 4848 years.years. Control is so eefficientfficient thatthat itit is is di difficultfficult to to find find a galleda galled stone stone fruit fruit tree, tree, when when previously, previously, crown crown gall had gall been had abeen major a problem.major problem. This paper This explainspaper explains how it works how andit works why onlyand pathogenswhy only arepathog inhibited.ens are A inhibited. commercial A biopesticidecommercial biopesticide is available inis Australia,available in Canada, Australia, Chile, Canada, New Zealand,Chile, New Turkey, Zealand, the USA, Turkey, South the Africa USA, andSouth Japan. Africa The and challenges Japan. The of commercialisingchallenges of commercialising a biopesticide area biopesticide outlined. Rigid -

Revised Taxonomy of the Family Rhizobiaceae, and Phylogeny of Mesorhizobia Nodulating Glycyrrhiza Spp
Division of Microbiology and Biotechnology Department of Food and Environmental Sciences University of Helsinki Finland Revised taxonomy of the family Rhizobiaceae, and phylogeny of mesorhizobia nodulating Glycyrrhiza spp. Seyed Abdollah Mousavi Academic Dissertation To be presented, with the permission of the Faculty of Agriculture and Forestry of the University of Helsinki, for public examination in lecture hall 3, Viikki building B, Latokartanonkaari 7, on the 20th of May 2016, at 12 o’clock noon. Helsinki 2016 Supervisor: Professor Kristina Lindström Department of Environmental Sciences University of Helsinki, Finland Pre-examiners: Professor Jaakko Hyvönen Department of Biosciences University of Helsinki, Finland Associate Professor Chang Fu Tian State Key Laboratory of Agrobiotechnology College of Biological Sciences China Agricultural University, China Opponent: Professor J. Peter W. Young Department of Biology University of York, England Cover photo by Kristina Lindström Dissertationes Schola Doctoralis Scientiae Circumiectalis, Alimentariae, Biologicae ISSN 2342-5423 (print) ISSN 2342-5431 (online) ISBN 978-951-51-2111-0 (paperback) ISBN 978-951-51-2112-7 (PDF) Electronic version available at http://ethesis.helsinki.fi/ Unigrafia Helsinki 2016 2 ABSTRACT Studies of the taxonomy of bacteria were initiated in the last quarter of the 19th century when bacteria were classified in six genera placed in four tribes based on their morphological appearance. Since then the taxonomy of bacteria has been revolutionized several times. At present, 30 phyla belong to the domain “Bacteria”, which includes over 9600 species. Unlike many eukaryotes, bacteria lack complex morphological characters and practically phylogenetically informative fossils. It is partly due to these reasons that bacterial taxonomy is complicated. -
Immunostrips® Validation Report
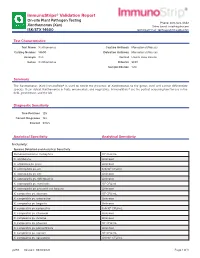
ImmunoStrips® Validation Report On-site Plant Pathogen Testing Phone: 800-622-4342 Xanthomonas (Xan) Sales Email: [email protected] ISK/STX 14600 Technical Email: [email protected] Test Characteristics Test Name Xanthomonas Capture Antibody Monoclonal (Mouse) Catalog Number 14600 Detection Antibody Monoclonal (Mouse) Acronym Xan Format Lateral Flow Device Genus Xanthomonas Diluents SEB1 Sample Dilution 1:20 Summary The Xanthomonas (Xan) ImmunoStrip® is used to detect the presence of Xanthomonas to the genus level and cannot differentiate species. It can detect Xanthomonas in fruits, ornamentals, and vegetables. ImmunoStrips® are the perfect screening tool for use in the field, greenhouse, and the lab. Diagnostic Sensitivity True Positives 125 Correct Diagnoses 122 Percent 97.6% Analytical Specificity Analytical Sensitivity Inclusivity: Species Detected and Analytical Sensitivity Stenotrophomonas maltophilia 106 CFU/mL X. albilibeans Unknown X. arboricola pv. pruni Unknown X. axonopodis pv. alii 6.9x104 CFU/mL X. axonopodis pv. citri Unknown X. axonopodis pv. diffenbachia Unknown X. axonopodis pv. manihotis 106 CFU/mL X. axonopodis pv. phaseoli var. fuscans Unknown X. campestris pv. aberrans 105 CFU/mL X. campestris pv. armoraciae Unknown X. campestris pv. begonia Unknown X. campestris pv. campestris 6.4x104 CFU/mL X. campestris pv. citromelo Unknown X. campestris pv. incanae Unknown X. campestris pv. phaseoli 105 CFU/mL X. campestris pv. poinsettiicola Unknown X. campestris pv. raphani 105 CFU/mL X. campestris pv. vesicatoria 4.0x105 CFU/mL p293 Revised: 08/18/2021 Page 1 of 3 Species Detected and Analytical Sensitivity X. campestris pv. vitians 105 CFU/mL X. campestris pv. zinnia 107 CFU/mL X. cannabis 1.6x104 CFU/mL X. -

Plant Root 11:33-39
www.plantroot.org 33 Original research article Biolistic-mediated plasmid-free transformation for induction of hairy roots Revi einw a tobaccorticle plantsShort report Gulnar Yasybaeva, Zilya Vershinina, Bulat Kuluev, Elena Mikhaylova, Andrey Baymiev and Aleksey Chemeris Institute of Biochemistry and Genetics, Ufa Scientific Centre, Russian Academy of Sciences (IBG USC RAS), pr. Oktyabrya 71, 450054, Ufa, Russia Corresponding author. G. Yasybaeva, E-mail: [email protected], Phone: +7-937-480-9956 Received on December 13, 2016; Accepted on March 8, 2017 Abstract: T-DNA of Ri-plasmid from Agrobacteri- transformed plant tissue, specifically, promotes the um rhizogenes is able to trigger the hairy root syn- appearance of hairy roots. Hairy roots culture (HRC) drome in infected plants. This natural phenomenon can be used for production of various secondary is used to generate hairy root cultures predomi- metabolites such as alkaloids, terpenoids, phenols, nantly only in dicotyledonous plants. We propose a glycosides (Sharifi et al. 2014). Some of them are a new method of hairy roots induction without Agro- source of pharmaceutical raw materials or other bacterium-mediated transformation. The 5461 bp economically valuable compounds. HRCs also can T-DNA region from A. rhizogenes A4 strain with all be used as recombinant proteins producers (Ono and four rol genes was amplified using primers contain- Tian 2011). ing sequences for left and right T-DNA borders on In natural conditions soil bacteria A. rhizogenes their 3’-ends. This amplicon was used for direct infects healthy plants by penetrating through injured transformation of tobacco leaf discs without A. rhi- roots. Thereby simple way of making hairy roots is zogenes and binary vectors. -

2010.-Hungria-MLI.Pdf
Mohammad Saghir Khan l Almas Zaidi Javed Musarrat Editors Microbes for Legume Improvement SpringerWienNewYork Editors Dr. Mohammad Saghir Khan Dr. Almas Zaidi Aligarh Muslim University Aligarh Muslim University Fac. Agricultural Sciences Fac. Agricultural Sciences Dept. Agricultural Microbiology Dept. Agricultural Microbiology 202002 Aligarh 202002 Aligarh India India [email protected] [email protected] Prof. Dr. Javed Musarrat Aligarh Muslim University Fac. Agricultural Sciences Dept. Agricultural Microbiology 202002 Aligarh India [email protected] This work is subject to copyright. All rights are reserved, whether the whole or part of the material is concerned, specifically those of translation, reprinting, re-use of illustrations, broadcasting, reproduction by photocopying machines or similar means, and storage in data banks. Product Liability: The publisher can give no guarantee for all the information contained in this book. The use of registered names, trademarks, etc. in this publication does not imply, even in the absence of a specific statement, that such names are exempt from the relevant protective laws and regulations and therefore free for general use. # 2010 Springer-Verlag/Wien Printed in Germany SpringerWienNewYork is a part of Springer Science+Business Media springer.at Typesetting: SPI, Pondicherry, India Printed on acid-free and chlorine-free bleached paper SPIN: 12711161 With 23 (partly coloured) Figures Library of Congress Control Number: 2010931546 ISBN 978-3-211-99752-9 e-ISBN 978-3-211-99753-6 DOI 10.1007/978-3-211-99753-6 SpringerWienNewYork Preface The farmer folks around the world are facing acute problems in providing plants with required nutrients due to inadequate supply of raw materials, poor storage quality, indiscriminate uses and unaffordable hike in the costs of synthetic chemical fertilizers. -

Gall-ID: Tools for Genotyping Gall-Causing Phytopathogenic Bacteria
Gall-ID: tools for genotyping gall-causing phytopathogenic bacteria Edward W. Davis II1,2,*, Alexandra J. Weisberg1,*, Javier F. Tabima1, Niklaus J. Grunwald1,2,3,4 and Jeff H. Chang1,2,3 1 Department of Botany and Plant Pathology, Oregon State University, Corvallis, OR, United States 2 Molecular and Cellular Biology Program, Oregon State University, Corvallis, OR, United States 3 Center for Genome Research and Biocomputing, Oregon State University, Corvallis, OR, United States 4 Horticultural Crops Research Laboratory, USDA-ARS, Corvallis, OR, United States * These authors contributed equally to this work. ABSTRACT Understanding the population structure and genetic diversity of plant pathogens, as well as the effect of agricultural practices on pathogen evolution, is important for disease management. Developments in molecular methods have contributed to increase the resolution for accurate pathogen identification, but those based on analysis of DNA sequences can be less straightforward to use. To address this, we developed Gall-ID, a web-based platform that uses DNA sequence information from 16S rDNA, multilocus sequence analysis and whole genome sequences to group disease-associated bacteria to their taxonomic units. Gall-ID was developed with a particular focus on gall-forming bacteria belonging to Agrobacterium, Pseudomonas savastanoi, Pantoea agglomerans, and Rhodococcus. Members of these groups of bacteria cause growth deformation of plants, and some are capable of infecting many species of field, orchard, and nursery crops. Gall-ID also enables the use of high-throughput sequencing reads to search for evidence for homologs of characterized virulence genes, and provides downloadable software pipelines for automating multilocus sequence analysis, analyzing genome sequences for average nucleotide identity, and constructing core genome phylogenies. -

Introduction to Plant Pathology {Richard N. Strange
Introduction to Plant Pathology Introduction to Plant Pathology Richard N. Strange University College London Copyright ß 2003 by John Wiley & Sons Ltd, The Atrium, Southern Gate, Chichester, West Sussex PO19 8SQ, England National l01243 779777 International (þ44) 1243 779777 e-mail (for orders and customer service enquiries): [email protected] Visit our Home Page on http://www.wiley.co.uk or http://www.wiley.com All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording, scanning or otherwise, except under the terms of the Copyright, Designs and Patents Act 1988 or under the terms of a licence issued by the Copyright Licensing Agency, 90 Tottenham Court Road, London, UK W1P 9 HE, without the permission in writing of the publisher. Other Wiley Editorial Offices John Wiley & Sons, Inc., 605 Third Avenue, New York, NY 10158-0012, USA Wiley-VCH Verlag GmbH, Pappelallee 3, D-69469 Weinheim, Germany John Wiley & Sons (Australia) Ltd, 33 Park Road, Milton, Queensland 4064, Australia John Wiley & Sons (Asia) Pte Ltd, 2 Clementi Loop #02-01, Jin Xing Distripark, Singapore 0512 John Wiley & Sons (Canada) Ltd, 22 Worcester Road, Rexdale, Ontario M9W 1L1, Canada Wiley also publishes its books in a variety of electronic formats. Some content that appears in print may not be available in electronic books. Library of Congress Cataloging-in-Publication Data Strange, Richard N. Introduction to plant pathology / Richard N. Strange. p. cm. Includes bibliographical references and index. ISBN 0-470-84972-X (Cloth : alk. -

A Review of Root Mat and Crown Gall Diseases to Inform Research on Control of Tomato Root Mat
Protected tomato – A review of root mat and crown gall diseases to inform research on control of tomato root mat Tim O’Neill and Sarah Mayne, ADAS John Elphinstone, Fera May 2016 PE 029 Contents Background ........................................................................................................................... 1 Summary .............................................................................................................................. 2 Technical review ................................................................................................................... 5 1. Occurrence of root mat ........................................................................................... 5 a) Rosaceous crops ................................................................................................. 6 b) Cucumber ............................................................................................................ 8 c) Tomato .............................................................................................................. 10 d) Other protected edible crops ............................................................................. 12 2. Occurrence of crown gall ...................................................................................... 12 a) Rosaceous crop ................................................................................................ 13 b) Other plant families ........................................................................................... 14 3. -

Rhizobium Tumorigenes Sp. Nov., a Novel Plant Tumorigenic Bacterium
www.nature.com/scientificreports OPEN Rhizobium tumorigenes sp. nov., a novel plant tumorigenic bacterium isolated from cane gall tumors on Received: 24 October 2017 Accepted: 4 June 2018 thornless blackberry Published: xx xx xxxx Nemanja Kuzmanović 1, Kornelia Smalla1, Sabine Gronow2 & Joanna Puławska 3 Four plant tumorigenic strains 932, 1019, 1078T and 1081 isolated from cane gall tumors on thornless blackberry (Rubus sp.) were characterized. They shared low sequence identity with related Rhizobium spp. based on comparisons of 16S rRNA gene (≤98%) and housekeeping genes atpD, recA and rpoB (<90%). Phylogenetic analysis indicated that the strains studied represent a novel species within the genus Rhizobium, with Rhizobium tubonense CCBAU 85046T as their closest relative. Furthermore, obtained average nucleotide identity (ANI) and in silico DNA–DNA hybridization (DDH) values calculated for whole- genome sequences of strain 1078T and related Rhizobium spp. confrmed the authenticity of the novel species. The ANI-Blast (ANIb), ANI-MUMmer (ANIm) and in silico DDH values between strain 1078T and most closely related R. tubonense CCBAU 85046T were 76.17%, 84.11% and 21.3%, respectively. The novel species can be distinguished from R. tubonense based on phenotypic and chemotaxonomic properties. Here, we demonstrated that four strains studied represent a novel species of the genus Rhizobium, for which the name Rhizobium tumorigenes sp. nov. is proposed (type strain 1078T = DSM 104880T = CFBP 8567T). R. tumorigenes is a new plant tumorigenic species carrying the tumor-inducing (Ti) plasmid. Plant tumorigenic bacteria belonging to the family Rhizobiaceae are associated with crown gall and cane gall diseases that can afect various plants1–3. -

Rhizobium As a Crop Enhancer and Biofertilizer for Increased Cereal Production
African Journal of Biotechnology Vol. 9(37), pp. 6001-6009, 13 September, 2010 Available online at http://www.academicjournals.org/AJB ISSN 1684–5315 © 2010 Academic Journals Review Rhizobium as a crop enhancer and biofertilizer for increased cereal production M. A. Baset Mia1,2 and Z. H. Shamsuddin1* 1Department of Land Management, Faculty of Agriculture, University Putra Malaysia, 43400 UPM, Serdang, Selangor DE, Malaysia. 2Department of Crop Botany, Faculty of Agriculture, Bangabandhu Sheikh Mujibur Rahman Agricultural University, Gazipur-1706, Bangladesh. Accepted 10 March, 2010 Greater production of cereals brings forth higher production cost and pollutes the soil environment due to excessive use of chemical fertilizers. Therefore, crop scientists are exploring an alternative source namely biofertilizers which are cost effective and environment friendly. In the biofertilizer technology, Rhizobium-legume is most common and widely used in different countries. Recently, it is also found that rhizobia can make an association with graminaceous plants such as rice, wheat, maize, barley millets and other cereals some time as endophytic without forming any nodule-like structure or causing any disease symptoms. Increasing the ability of rhizobia in biofertilizer, crop enhancing activity in non- legumes especially cereal grains would be a useful technology for increased crop yields among resource-poor farmers. Recent findings showed both more crop enhancing and biofertilizer attributes in cereal crops due to rhizobial inoculation. In addition, plant nutrients like P, K, Ca, Mg and even Fe accumulation were also observed. Therefore, further research in this area will be able to develop a sustainable biofertilizer technology for greater and environment friendly cereal production system. -
Role of Plasmids in Plant-Bacteria Interactions
Plasmids in Plant-Bacteria Interactions Schierstaedt et al. Curr. Issues Mol. Biol. (2019) 30: 17-38. caister.com/cimb Role of Plasmids in Plant-Bacteria Interactions Jasper Schierstaedt1, Nina Bziuk2, Nemanja bacteria interactions. Furthermore, we discuss tools Kuzmanović2, Khald Blau2, Kornelia Smalla2 and available to study the plant-associated mobilome, its Sven Jechalke3* transferability, and its bacterial hosts. 1Leibniz Institute of Vegetable and Ornamental Introduction Crops (IGZ), Department Plant-microbe systems, Plant-associated microorganisms are considered to Theodor-Echtermeyer-Weg 1, 14979 Großbeeren, be of great importance for plant health, plant Germany productivity and ecosystem functioning. They 2Julius Kühn-Institut - Federal Research Centre for expand the metabolic repertoire of plants, increase Cultivated Plants (JKI), Institute for Epidemiology the resource uptake and provide novel nutritional and Pathogen Diagnostics, Messeweg 11-12, 38104 and defense pathways (Berendsen et al., 2012; Braunschweig, Germany Berg et al., 2014). Therefore, the genetic 3Justus Liebig University Giessen, Institute for information provided by the plant microbiome is also Phytopathology, Heinrich-Buff-Ring 26-32, 35392 called the second genome of the plant (Berendsen Gießen, Germany et al., 2012). In the phytosphere, mutualistic associations were studied in great detail for rhizobia *[email protected] and mycorrhizae, rhizobacteria with plant growth promoting or biocontrol activity. However, also DOI: https://dx.doi.org/10.21775/cimb.030.017 parasitic interactions with plant pathogens are well- studied today. Plants are able to influence soil Abstract properties, e.g. by the release of nutrients and Plants are colonized by diverse microorganisms, secondary metabolites via root exudation, which are which may positively or negatively influence the used to combat pathogenic microorganisms while plant fitness.