Bridion, INN-Sugammadex
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

12-6202 Document: 01018895054 Date Filed: 08/10/2012 Page: 1 FILED United States Court of Appeals UNITED STATES COURT of APPEALS Tenth Circuit
Appellate Case: 12-6202 Document: 01018895054 Date Filed: 08/10/2012 Page: 1 FILED United States Court of Appeals UNITED STATES COURT OF APPEALS Tenth Circuit FOR THE TENTH CIRCUIT August 10, 2012 Elisabeth A. Shumaker Clerk of Court MICHAEL EDWARD HOOPER, Plaintiff-Appellant, v. No. 12-6202 (D.C. No. 5:12-cv-00758-M) JUSTIN JONES, Director DOC; (W.D. Okla.) RANDALL G. WORKMAN, Warden; DOES, Unknown Executioners, Defendants-Appellees. ORDER AND JUDGMENT* Before MURPHY, O’BRIEN, and HOLMES, Circuit Judges. Michael Edward Hooper, an Oklahoma state prisoner scheduled for execution by lethal injection on August 14, 2012, appeals from the district court’s order * After examining the briefs and appellate record, this panel has determined unanimously that oral argument would not materially assist the determination of this appeal. See Fed. R. App. P. 34(a)(2); 10th Cir. R. 34.1(G). The case is therefore ordered submitted without oral argument. This order and judgment is not binding precedent, except under the doctrines of law of the case, res judicata, and collateral estoppel. It may be cited, however, for its persuasive value consistent with Fed. R. App. P. 32.1 and 10th Cir. R. 32.1. Appellate Case: 12-6202 Document: 01018895054 Date Filed: 08/10/2012 Page: 2 denying his motion for a preliminary injunction seeking to stay his execution. Exercising jurisdiction under 28 U.S.C. § 1292(a)(1), we AFFIRM.1 I Mr. Hooper was tried and convicted on three counts of first-degree murder and sentenced to death. See Hooper v. State, 947 P.2d 1090 (Okla. -

Quantitative Analysis of the Aminosteroidal Muscle Relaxant
J. Ecophysiol. Occup. Hlth. 3 & 4 (2013) 29-37 ® 2013 The Academy of Environmental Biology, India Quantitative Analysis of the Aminosteroidal Muscle Relaxant Vecuronium in Rat Liver and Kidneys using Ion-pairing and Liquid Chromatography-Tandem Mass Spectrometry Risha Jasmine Nathan*, P. Sharma, Lily Saroj Nathan1 Forensic Chemistry and Toxicology, Lok Nayak Jayaprakash Narayan National Institute of Criminology, New Delhi 1Ewing Christian College, University of Allahabad Abstract : Muscle relaxants are occasionally encountered in forensic toxicological cases for identification and quantitation. Due to unavailability of human samples, the analysis of the muscle relaxant vecuronium was done using post-mortem rat visceral samples. Rats were injected with bolus overdose of the drug and the post mortem samples were subject to ion-pair extraction with metanil yellow dye. Method for identification of the drug by LC-MS-MS was established first with secondary standard aqueous vecuronium solutions and then applied on the questioned samples. Quantitation was done to find the quantity of the drug in liver and kidney samples. Keywords : Aminosteroid, Muscle relaxant Vecuronium, Liquid Chromatography-Tandem Mass Spectrometry. Introduction ventilation is made available to the patient so as Muscle relaxants, better known as neuro- to maintain respiration as these drugs may also muscular-blocking drugs, are a class of drugs paralyze the diaphragm at the therapeutic dose that block neuromuscular transmission at required (Dripps, 1959). Despite taking neuromuscular junction which causes paralysis precautions, neuromuscular blocking drugs of the affected skeletal muscles. Of the two have been reported to have inadvertently classes, non-depolarizing neuromuscular caused deaths of patients by accident agents work without depolarizing the motor end (Newsletters, Nurse Advise, 2006). -

Product Monograph Vecuronium Bromide For
PRODUCT MONOGRAPH PrVECURONIUM BROMIDE FOR INJECTION Non-depolarizing Skeletal Neuromuscular Blocking Agent Pharmaceutical Partners of Canada Inc. Date of Preparation: 45 Vogell Road, Suite 200 January 15, 2008 Richmond Hill, ON L4B 3P6 Control No.: 119276 PRODUCT MONOGRAPH PrVECURONIUM BROMIDE FOR INJECTION Non-depolarizing Skeletal Neuromuscular Blocking Agent THIS DRUG SHOULD BE ADMINISTERED ONLY BY ADEQUATELY TRAINED INDIVIDUALS FAMILIAR WITH ITS ACTIONS, CHARACTERISTICS AND HAZARDS ACTIONS AND CLINICAL PHARMACOLOGY Vecuronium Bromide for Injection is a non-depolarizing neuromuscular blocking agent of intermediate duration possessing all of the characteristic pharmacological actions of this class of drugs (curariform). It acts by competing for cholinergic receptors at the motor end-plate. The antagonism to acetylcholine is inhibited and neuromuscular block reversed by acetylcholinesterase inhibitors such as neostigmine. Vecuronium Bromide for Injection is about 1/3 more potent than pancuronium; the duration of neuromuscular blockade produced by Vecuronium Bromide for Injection is shorter than that of pancuronium at initially equipotent doses. The time to onset of paralysis decreases and the duration of maximum effect increases with increasing Vecuronium Bromide for Injection doses. The use of a peripheral nerve stimulator is of benefit in assessing the degree of muscular relaxation. The ED90 (dose required to produce 90% suppression of the muscle twitch response with balanced anesthesia) has averaged 0.057 mg/kg (0.049 to 0.062 mg/kg in various studies). An initial Vecuronium Bromide for Injection dose of 0.08 to 0.10 mg/kg generally produces first depression of twitch in approximately 1 minute, good or excellent intubation conditions within 2.5 to 3 minutes, and maximum neuromuscular blockade within 3 to 5 minutes of injection in Vecuronium Bromide for Injection, Product Monograph Page 2 of 28 most patients. -
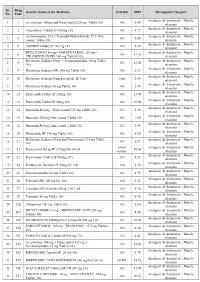
PMBJP Product.Pdf
Sr. Drug Generic Name of the Medicine Unit Size MRP Therapeutic Category No. Code Analgesic & Antipyretic / Muscle 1 1 Aceclofenac 100mg and Paracetamol 325 mg Tablet 10's 10's 8.00 relaxants Analgesic & Antipyretic / Muscle 2 2 Aceclofenac Tablets IP 100mg 10's 10's 4.37 relaxants Acetaminophen 325 + Tramadol Hydrochloride 37.5 film Analgesic & Antipyretic / Muscle 3 4 10's 8.00 coated Tablet 10's relaxants Analgesic & Antipyretic / Muscle 4 5 ASPIRIN Tablets IP 150 mg 14's 14's 2.70 relaxants DICLOFENAC 50 mg+ PARACETAMOL 325 mg+ Analgesic & Antipyretic / Muscle 5 6 10's 11.30 CHLORZOXAZONE 500 mg Tablets 10's relaxants Diclofenac Sodium 50mg + Serratiopeptidase 10mg Tablet Analgesic & Antipyretic / Muscle 6 8 10's 12.00 10's relaxants Analgesic & Antipyretic / Muscle 7 9 Diclofenac Sodium (SR) 100 mg Tablet 10's 10's 6.12 relaxants Analgesic & Antipyretic / Muscle 8 10 Diclofenac Sodium 25mg per ml Inj. IP 3 ml 3 ml 2.00 relaxants Analgesic & Antipyretic / Muscle 9 11 Diclofenac Sodium 50 mg Tablet 10's 10's 2.90 relaxants Analgesic & Antipyretic / Muscle 10 12 Etoricoxilb Tablets IP 120mg 10's 10's 33.00 relaxants Analgesic & Antipyretic / Muscle 11 13 Etoricoxilb Tablets IP 90mg 10's 10's 25.00 relaxants Analgesic & Antipyretic / Muscle 12 14 Ibuprofen 400 mg + Paracetamol 325 mg Tablet 10's 15's 5.50 relaxants Analgesic & Antipyretic / Muscle 13 15 Ibuprofen 200 mg film coated Tablet 10's 10's 1.80 relaxants Analgesic & Antipyretic / Muscle 14 16 Ibuprofen 400 mg film coated Tablet 10's 15's 3.50 relaxants Analgesic & Antipyretic -

Vecuronium Bromide As Control
A COMPARATIVE STUDY TO FIND AN IDEAL INTUBATING DOSE OF INJ. ROCURONIUM BROMIDE USING INJ. VECURONIUM BROMIDE AS CONTROL. Dissertation submitted In partial fulfillment for the award of M.D DEGREE EXAMINATION M.D. ANAESTHESIOLOGY & CRITICAL CARE BRANCH X GOVT KILPAUK MEDICAL COLLEGE AND HOSPITAL SUBMITTED TO THE TAMILNADU DR. MGR MEDICAL UNIVERSITY CHENNAI APRIL – 2011 1 DECLARATION I, Dr. D.Shunmuga Priya, solemnly declare that the dissertation, “A COMPARATIVE STUDY TO FIND AN IDEAL INTUBATING DOSE OF INJ. ROCURONIUM BROMIDE USING INJ. VECURONIUM BROMIDE AS CONTROL” is a bonafide work done by me in the Department of Anaesthesiology and Critical Care, Government Kilpauk, Medical College, Chennai under the able guidance of Prof. Dr. P.S. Shanmugam, MD., DA., Professor and HOD, Department of Anaesthesiology and Critical Care, Government Kilpauk Medical College, Chennai. Place : Chennai Date : (Dr. D.SHUNMUGA PRIYA) 5 CERTIFICATE This is to certify that this dissertation titled “A COMPARATIVE STUDY TO FIND AN IDEAL INTUBATING DOSE OF INJ. ROCURONIUM BROMIDE USING INJ. VECURONIUM BROMIDE AS CONTROL” has been prepared by Dr. D.SHUNMUGA PRIYA under my supervision in the Department of Anaesthesiology and Critical Care, Government Kilpauk Medical College, Chennai during the academic period 2008-2011 and is being submitted to the Tamil Nadu Dr. M.G.R. Medical University, Chennai in partial fulfillment of the University regulation for the award of the Degree of Doctor of Medicine (MD Anaesthesiology and Critical Care) and her dissertation is a bonafide work. Prof..Dr. V.Kanagasabai, M.D Prof. Dr. P.S.Shanmugam, M.D., D.A. DEAN Professor & HOD, Government Kilpauk Medical College Department of Anaesthesiology and & Hospital, Critical Care, Chennai. -

Vecureinj.Pdf
NEW ZEALAND DATA SHEET VECURE 1. Product Name VECURE 10 mg, powder for injection. 2. Qualitative and Quantitative Composition VECURE 10 mg, 1 vial contains 10 mg vecuronium bromide. When reconstituted with 5 mL water for injection corresponds to 2 mg vecuronium bromide per mL in a clear almost clear isotonic solution. For the full list of excipients, see section 6.1 3. Pharmaceutical Form White, freeze dried powder for injection. Following reconstitution with solvent (water for injection) the resultant solution is isotonic and has a pH of 4. 4. Clinical Particulars 4.1 Therapeutic indications Vecure is indicated as an adjunct to general anaesthesia to facilitate tracheal intubation and to provide skeletal muscle relaxation during surgery. 4.2 Dose and method of administration Dosage Like other neuromuscular blocking agents, VECURE should only be administered by, or under supervision of, experienced clinicians who are familiar with the action and use of these agents. Like with other neuromuscular blocking agents, the dosage of VECURE should be individualised in each patient. The anaesthetic method used, the expected duration of surgery, the possible interaction with other medicines that are administered before or during anaesthesia and the condition of the patient should be taken into account when determining the dose. The use of an appropriate neuromuscular monitoring technique is recommended to monitor neuromuscular block and recovery. Inhalational anaesthetics do potentiate the neuromuscular blocking effects of VECURE. This potentiation however, becomes clinically relevant in the course of anaesthesia, when the volatile agents have reached the tissue concentrations required for this interaction. Consequently, adjustments with VECURE should be made by administering smaller maintenance doses at less frequent intervals or by using lower infusion rates of VECURE during long lasting procedures (longer than 1 hour) under inhalational anaesthesia (see section 4.5). -

COMPARISON of ROCURONIUM BROMIDE and SUCCINYLCHOLINE CHLORIDE for USE DURING RAPID SEQUENCE INTUBATION in ADULTS Ch
DOI: 10.18410/jebmh/2015/672 ORIGINAL ARTICLE COMPARISON OF ROCURONIUM BROMIDE AND SUCCINYLCHOLINE CHLORIDE FOR USE DURING RAPID SEQUENCE INTUBATION IN ADULTS Ch. Penchalaiah1, N. Jagadeesh Babu2, T. Kumar3 HOW TO CITE THIS ARTICLE: Ch. Penchalaiah, N. Jagadeesh Babu, T. Kumar. ”Comparison of Rocuronium Bromide and Succinylcholine Chloride for Use during Rapid Sequence Intubation in Adults”. Journal of Evidence based Medicine and Healthcare; Volume 2, Issue 32, August 10, 2015; Page: 4796-4806, DOI: 10.18410/jebmh/2015/672 ABSTRACT: BACKGROUND AND OBJECTIVE: The goal of rapid sequence intubation is to secure the patients airway smoothly and quickly, minimizing the chances of regurgitation and aspiration of gastric contents. Traditionally succinylcholine chloride has been the neuromuscular blocking drug of choice for use in rapid sequence intubation because of its rapid onset of action and profound relaxation. Succinylcholine chloride remains unsurpassed in providing ideal intubating conditions. However the use of succinylcholine chloride is associated with many side effects like muscle pain, bradycardia, hyperkalaemia and rise in intragastric and intraocular pressure. Rocuronium bromide is the only drug currently available which has the rapidity of onset of action like succinylcholine chloride. Hence the present study was undertaken to compare rocuronium bromide with succinylcholine chloride for use during rapid sequence intubation in adult patients. METHODOLOGY: The study population consisted of 90 patients aged between 18-60 years posted for various elective surgeries requiring general anaesthesia. Study population was randomly divided into 3 groups with 30 patients in each sub group. 1. Group I: Intubated with 1 mg kg-1 of succinylcholine chloride (n=30). -

1. NAME of the MEDICINAL PRODUCT Sugammadex
1. NAME OF THE MEDICINAL PRODUCT Sugammadex Pharmazac 100 mg/mL solution for injection 2. QUALITATIVE AND QUANTITATIVE COMPOSITION 1 mL contains sugammadex sodium equivalent to 100 mg sugammadex. Each vial of 2 mL contains sugammadex sodium equivalent to 200 mg sugammadex. Each vial of 5 mL contains sugammadex sodium equivalent to 500 mg sugammadex. Excipient(s) with known effect Each mL contains up to 9.5 mg sodium (see section 4.4). For the full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM Solution for injection (injection). Clear and colourless to slightly yellow/brown solution free from visible particles. The pH is between 7 and 8 and osmolality is between 300 and 500 mOsm/kg. 4. CLINICAL PARTICULARS 4.1 Therapeutic indications Reversal of neuromuscular blockade induced by rocuronium or vecuronium in adults. For the paediatric population: sugammadex is only recommended for routine reversal of rocuronium induced blockade in children and adolescents aged 2 to 17 years. 4.2 Posology and method of administration Posology Sugammadex should only be administered by, or under the supervision of an anaesthetist. The use of an appropriate neuromuscular monitoring technique is recommended to monitor the recovery of neuromuscular blockade (see section 4.4). The recommended dose of sugammadex depends on the level of neuromuscular blockade to be reversed. The recommended dose does not depend on the anaesthetic regimen. Sugammadex can be used to reverse different levels of rocuronium or vecuronium induced neuromuscular blockade: Adults Routine reversal: A dose of 4 mg/kg sugammadex is recommended if recovery has reached at least 1-2 post- tetanic counts (PTC) following rocuronium or vecuronium induced blockade. -

Reversal of Neuromuscular Bl.Pdf
JosephJoseph F.F. Answine,Answine, MDMD Staff Anesthesiologist Pinnacle Health Hospitals Harrisburg, PA Clinical Associate Professor of Anesthesiology Pennsylvania State University Hospital ReversalReversal ofof NeuromuscularNeuromuscular BlockadeBlockade DefiniDefinitionstions z ED95 - dose required to produce 95% suppression of the first twitch response. z 2xED95 – the ED95 multiplied by 2 / commonly used as the standard intubating dose for a NMBA. z T1 and T4 – first and fourth twitch heights (usually given as a % of the original twitch height). z Onset Time – end of injection of the NMBA to 95% T1 suppression. z Recovery Time – time from induction to 25% recovery of T1 (NMBAs are readily reversed with acetylcholinesterase inhibitors at this point). z Recovery Index – time from 25% to 75% T1. PharmacokineticsPharmacokinetics andand PharmacodynamicsPharmacodynamics z What is Pharmacokinetics? z The process by which a drug is absorbed, distributed, metabolized and eliminated by the body. z What is Pharmacodynamics? z The study of the action or effects of a drug on living organisms. Or, it is the study of the biochemical and physiological effects of drugs. For example; rocuronium reversibly binds to the post synaptic endplate, thereby, inhibiting the binding of acetylcholine. StructuralStructural ClassesClasses ofof NondepolarizingNondepolarizing RelaxantsRelaxants z Steroids: rocuronium bromide, vecuronium bromide, pancuronium bromide, pipecuronium bromide. z Benzylisoquinoliniums: atracurium besylate, mivacurium chloride, doxacurium chloride, cisatracurium besylate z Isoquinolones: curare, metocurine OnsetOnset ofof paralysisparalysis isis affectedaffected by:by: z Dose (relative to ED95) z Potency (number of molecules) z KEO (plasma equilibrium constant - chemistry/blood flow) — determined by factors that modify access to the neuromuscular junction such as cardiac output, distance of the muscle from the heart, and muscle blood flow (pharmacokinetic variables). -

Inhibitory Actions of Vecuronium Bromide on Acetylcholine and Glutamate Responses at the Frog and Crayfish Neuromuscular Junction
Inhibitory Actions of Vecuronium Bromide on Acetylcholine and Glutamate Responses at the Frog and Crayfish Neuromuscular Junction Haruhiko SHINOZAKI and Michiko ISHIDA The Tokyo Metropolitan Institute of Medical Science, 3-18-22 Honkomagome, Bunkyo-ku, Tokyo 113, Japan Accepted November 7, 1983 Abstract-Effects of vecuronium bromide, an analog of pancuronium, on the cholinergic and glutamatergic neuromuscular junction were investigated. Vecuronium depressed the postsynaptic response of the frog end-plate at lower concentrations than 10-6 g/ml without affecting the presynaptic events. Vecuronium decreased the amplitude of the double ACh potential, but the second potential was more markedly reduced than the first. In analogy with d-tubocurarine, this suggests that vecuronium may act in part as an open channel blocker at the frog end-plate. Vecuro nium depressed both the glutamate response and the excitatory junctional potential at the crayfish neuromuscular junction, although high concentrations were required. The drug increased the decay rate of extracellularly recorded excitatory functional potentials at the crayfish neuromuscular junction. The reduction of the crayfish synaptic response caused by vecuronium can be explained by the open channel blocking action at this functional site. The problem that cholinergic antagonists possess a property of channel blocking at the other transmitter system was discussed. Vecuronium is an analog of pancuronium, The neuromuscular blocking action of pan but is not the bis-quarternary ammonium, one curonium has been well documented (2). quarternary moiety of pancuronium being The comparison of pharmacological activities replaced by a tertiary amine (Fig. 11. Although of vecuronium to pancuronium was already it is known that replacement of one quar done in some earlier studies (3-6), and ternary moiety of the bis-quarternary am the effectiveness of both drugs is almost monium structure by a primary amine group the same, however, electrophysiological results in a considerable loss of potency (1), studies are lacking. -

Pdf Medication Assisted Induction Guidelines Final 05072019
Medication Assisted Induction (MAI) Regional Guidelines 2019 Edition Effective June 1, 2019 Lord Fairfax EMS Council, Inc. 180-1 Prosperity Drive Winchester, Virginia 22602 Phone 540-665-0014 Fax 540-722-0094 www.lfems.vaems.org © 2019 LFEMS Medication Assisted Induction (MAI) Guidelines – Revised 05/07/2019 1 LORD FAIRFAX EMERGENCY MEDICAL SERVICES COUNCIL, INC. MEDICATION ASSISTED INDUCTION (MAI) REGIONAL GUIDELINES TABLE OF CONTENTS MEDICATION ASSISTED INDUCTION (MAI) REGIONAL GUIDELINES REVISION HISTORY ........................................................................................................ 3 PREFACE .........................................................................................................................4 MAI AUTHORIZATION TO PRACTICE ............................................................................. 5 REGIONAL PROVIDER GUIDELINES ............................................................................. 6 FENTANYL (SUBLIMAZE®) ........................................................................................... 12 KETAMINE (KETALAR®) ............................................................................................... 13 MIDAZOLAM (VERSED®) .............................................................................................. 14 ROCURONIUM BROMIDE (ZEMURON)(®) ................................................................... 15 VECURONIUM BROMIDE (NORCURON)(®) ................................................................. 16 © 2019 LFEMS Medication Assisted -

Hikma Launches Vecuronium Bromide for Injection
Press Release Hikma launches Vecuronium Bromide for Injection London, April 3, 2020 – Hikma Pharmaceuticals PLC (Hikma, Group), the multinational generic pharmaceutical company, has launched Vecuronium Bromide for Injection, 10mg, in the United States through its US affiliate, Hikma Pharmaceuticals USA Inc.1 According to IQVIA, US sales of Vecuronium Bromide for Injection, 10mg, were approximately $6 million in the 12 months ending February 2020. Vecuronium Bromide for Injection is indicated as an adjunct to general anesthesia, to facilitate endotracheal intubation and to provide skeletal muscle relaxation during surgery or mechanical ventilation. Hikma is the third largest US supplier of generic injectable medicines by volume, with a growing portfolio of over 100 products. Today one in every six injectable generic medicines used in US hospitals is a Hikma product. - ENDS - Enquiries Hikma Pharmaceuticals PLC Susan Ringdal +44 (0)20 7399 2760/ +44 7776 477050 EVP, Strategic Planning and Global Affairs [email protected] Steve Weiss +1 732 720 2830/ +1 732 788 8279 David Belian +1 732 720 2814/+1 848 254 4875 US Communications and Public Affairs [email protected] About Hikma (LSE: HIK) (NASDAQ Dubai: HIK) (OTC: HKMPY) (rated Ba1/stable Moody's and BB+/positive S&P) Hikma helps put better health within reach every day for millions of people in more than 50 countries around the world. For more than 40 years, we've been creating high-quality medicines and making them accessible to the people who need them. Headquartered in the UK, we are a global company with a local presence across the United States (US), the Middle East and North Africa (MENA) and Europe, and we use our unique insight and expertise to transform cutting-edge science into innovative solutions that transform people's lives.