Co2 Storage As Carbonate Minerals
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mineralogy of the Martian Surface
EA42CH14-Ehlmann ARI 30 April 2014 7:21 Mineralogy of the Martian Surface Bethany L. Ehlmann1,2 and Christopher S. Edwards1 1Division of Geological & Planetary Sciences, California Institute of Technology, Pasadena, California 91125; email: [email protected], [email protected] 2Jet Propulsion Laboratory, California Institute of Technology, Pasadena, California 91109 Annu. Rev. Earth Planet. Sci. 2014. 42:291–315 Keywords First published online as a Review in Advance on Mars, composition, mineralogy, infrared spectroscopy, igneous processes, February 21, 2014 aqueous alteration The Annual Review of Earth and Planetary Sciences is online at earth.annualreviews.org Abstract This article’s doi: The past fifteen years of orbital infrared spectroscopy and in situ exploration 10.1146/annurev-earth-060313-055024 have led to a new understanding of the composition and history of Mars. Copyright c 2014 by Annual Reviews. Globally, Mars has a basaltic upper crust with regionally variable quanti- by California Institute of Technology on 06/09/14. For personal use only. All rights reserved ties of plagioclase, pyroxene, and olivine associated with distinctive terrains. Enrichments in olivine (>20%) are found around the largest basins and Annu. Rev. Earth Planet. Sci. 2014.42:291-315. Downloaded from www.annualreviews.org within late Noachian–early Hesperian lavas. Alkali volcanics are also locally present, pointing to regional differences in igneous processes. Many ma- terials from ancient Mars bear the mineralogic fingerprints of interaction with water. Clay minerals, found in exposures of Noachian crust across the globe, preserve widespread evidence for early weathering, hydrothermal, and diagenetic aqueous environments. Noachian and Hesperian sediments include paleolake deposits with clays, carbonates, sulfates, and chlorides that are more localized in extent. -

The Mineralogy of the British Permian Evaporites
460 The mineralogy of the British Permian evaporites By F. H. STEWART Department of Geology, University of Edinburgh Summary. The occurrence of the minerals is reviewed. Consideration of their genesis leads to a tentative list of primary minerals, and of early diagenetie, later diagenetic and geothermal metamorphic, and late near-surface changes. N 1943 Professor Tilley recorded the occurrence of po]yhalite in the I rocks of the D'Arcy Exploration Company's E.2 borehole at Aislaby, near Whitby; this was the first published record of a potassium-bearing salt in British evaporites. The occurrence of rocks containing sylvine (Lees and Taitt, 1946) prompted further exploration by Imperial Chemical Industries and Fisons Ltd. after the war, and this showed the presence of large deposits of potassium ores in the Whitby region. There is now a considerable literature related to these, and it seems timely to give a short review of the mineralogy of the British Permian evaporites. The writer is most grateful to Fisons Ltd. who have kindly allowed him to use unpublished information from his study of their boreholes F.1 at Robin Hood's Bay, F.2 at Staintondale, F.3 at Little Beck, and F.4 at Hawsker, in the Whitby Scarborough district. The position of these and the other boreholes mentioned in this paper can be found in the map of Dunham (1960). Dist~'ibution of the minerals The evaporites occur in two principal areas east and west of the Pennine Hills. In the western area (the Vale of Eden, west Cumberland, and the Furness district of Lancashire), where four evaporite beds have been recognized (Hollingworth, 1942), the mineralogy is relatively simple; carbonates, anhydrite, and gypsum are the main constituents. -

Crystallization of Kainite from Solutions in Syste
ineering ng & E P l r a o c i c e m s e s Journal of h T C e f c h o Kostiv and Basystiuk, J Chem Eng Process Technol 2016, 7:3 l ISSN: 2157-7048 n a o n l o r g u y o J Chemical Engineering & Process Technology DOI: 10.4172/2157-7048.1000298 Research Article Article OpenOpen Access Access + 2+ + - 2- Crystallization of Kainite from Solutions in System K , Mg , Na // Cl , SO4 -Н2О Kostiv IY1 and Yа І Basystiuk2* 1State Enterprise Scientific - Research Gallurgi Institute 5a, Fabrichna Str., 76000 Kalush, Ukraine 2Precarpathian National University named after V. Stephanyk 57, Shevchenko Str., 76025 Ivano-Frankivsk, Ukraine Abstract In isothermal conditions have been studied the influence of system solution sea salts during their evaporation and crystallization to composition of received liquid and solid phases. It is shown that evaporation of the solution leading 2- to its supersaturation by sulfate salts. In the liquid phase before crystallization of kainite concentration of SO4 ions increases to 10% and above. Through this probability of formation of crystals increases and the result of evaporation 2- is the formation of finely dispersed kainite. During crystallization of kainite concentration of SO4 in the liquid phase 2- 2+ decreases rapidly. Decreasing its intensity increases with increasing value k=E SO4 : E Mg of initial solution. For 2- the degree of evaporation of 20.0% and 31.0% concentration of SO4 in evaporated solution on the value of k does not change. Most forms of potassium salts in the solid phase for the value k=0.7573 and reaches a maximum value at 82% evaporation rate of 31%. -

Mechanism and Kinetics of Dehydration of Epsomite Crystals Formed in the Presence of Organic Additives
J. Phys. Chem. B 2007, 111, 41-52 41 Mechanism and Kinetics of Dehydration of Epsomite Crystals Formed in the Presence of Organic Additives Encarnacio´n Ruiz-Agudo,† J. Daniel Martı´n-Ramos,‡ and Carlos Rodriguez-Navarro* Departamento de Mineralogı´a y Petrologı´a, UniVersidad de Granada, FuentenueVa s/n, 18002 Granada, Spain ReceiVed: July 14, 2006; In Final Form: October 10, 2006 The thermal dehydration of epsomite (MgSO4‚7H2O) crystals grown in the presence and absence of organic additives (phosphonates, carboxylic acids, and polyacrylic acid derivatives) was studied by means of thermogravimetry (TG), differential scanning calorimetry (DSC), X-ray thermodiffraction (XRTD), and environmental scanning electron microscopy (ESEM). In situ XRTD analyses (in air, 30% relative humidity) show an epsomite f hexahydrite (MgSO4‚6H2O) transition at 25-38 °C, followed by formation of amorphous phase(s) at T > 43-48 °C, and MgSO4 crystallization at ∼300 °C. Kinetic parameters (ER and A) were determined for the main dehydration step (25-160 °C), which corresponds to a MgSO4‚7H2O f MgSO4‚ H2O transition, by applying two isoconversional methods to nonisothermal TG data obtained at different heating rates (â ) 1, 3, and 5 K‚min-1). In situ, hot-stage ESEM observations of the thermal dehydration of epsomite crystals are consistent with the nonisothermal kinetic study and, along with XRTD results, allow us to propose a dehydration mechanism which includes an early nucleation and growth event, followed by the advancement of the reaction interface (3D phase boundary reaction). Both ER and A values increase in the presence of the most effective crystallization inhibitors tested. -

Stability of Magnesium Sulfate Minerals in Martian Environments
Lunar and Planetary Science XXXVI (2005) 2290.pdf STABILITY OF MAGNESIUM SULFATE MINERALS IN MARTIAN ENVIRONMENTS. G.M. Marion1 and J.S. Kargel2, 1Desert Research Institute, 2215 Raggio Parkway, Reno, NV 89512, [email protected], 2U.S. Geological Survey, 2255 N. Gemini Dr., Flagstaff, AZ 86001, [email protected]. Introduction: Viking Lander, Pathfinder, and –3.6°C for a pure MgSO4-H2O system. In this system, Mars Exploration Rover missions to Mars have found the lower hydrates, hexahydrite and kieserite, only abundant sulfur in surface soils and rocks, and the best form at higher temperatures (48°C and 69°C, respec- indications are that magnesium sulfates are among the tively). key hosts [1-6]. At Meridiani Planum, MgSO4 salts The presence of other constituents in ternary (or constitute 15 to 40 wt.% of sedimentary rocks [3-5,7]. higher) systems can lower the temperatures at which Additional S is hosted by gypsum and jarosite. Reflec- lower hydrates are stable. For example, hexahydrite is tance and thermal emission spectroscopy is consistent stable at 25°C in a MgSO4-MgCl2-H2O system at 3.8 with the presence of kieserite (MgSO4•H2O) and ep- m MgCl2 [12]. This requires high chloride concentra- somite (MgSO4•7H2O) [3]. Theoretically, the tions that may occur in Martian environments that dodecahydrate (MgSO4•12H2O) should also have have previously undergone extensive sulfate precipita- precipitated [8]. tion and chloride concentration due to extreme freez- We first examine theoretically which MgSO4 min- ing or desiccating conditions [3-5]. erals should have precipitated on Mars, and then how Based on the Rover mission to Meridiani Planum, dehydration might have altered these minerals. -

High Temperature Sulfate Minerals Forming on the Burning Coal Dumps from Upper Silesia, Poland
minerals Article High Temperature Sulfate Minerals Forming on the Burning Coal Dumps from Upper Silesia, Poland Jan Parafiniuk * and Rafał Siuda Faculty of Geology, University of Warsaw, Zwirki˙ i Wigury 93, 02-089 Warszawa, Poland; [email protected] * Correspondence: j.parafi[email protected] Abstract: The subject of this work is the assemblage of anhydrous sulfate minerals formed on burning coal-heaps. Three burning heaps located in the Upper Silesian coal basin in Czerwionka-Leszczyny, Radlin and Rydułtowy near Rybnik were selected for the research. The occurrence of godovikovite, millosevichite, steklite and an unnamed MgSO4, sometimes accompanied by subordinate admixtures of mikasaite, sabieite, efremovite, langbeinite and aphthitalite has been recorded from these locations. Occasionally they form monomineral aggregates, but usually occur as mixtures practically impossible to separate. The minerals form microcrystalline masses with a characteristic vesicular structure resembling a solidified foam or pumice. The sulfates crystallize from hot fire gases, similar to high temperature volcanic exhalations. The gases transport volatile components from the center of the fire but their chemical compositions are not yet known. Their cooling in the near-surface part of the heap results in condensation from the vapors as viscous liquid mass, from which the investigated minerals then crystallize. Their crystallization temperatures can be estimated from direct measurements of the temperatures of sulfate accumulation in the burning dumps and studies of their thermal ◦ decomposition. Millosevichite and steklite crystallize in the temperature range of 510–650 C, MgSO4 Citation: Parafiniuk, J.; Siuda, R. forms at 510–600 ◦C and godovikovite in the slightly lower range of 280–450 (546) ◦C. -
![The Paragenesis of Sylvine, Carnallite, Polyhalite, and ]Cieserite in Eskdale Borings Nos](https://docslib.b-cdn.net/cover/6717/the-paragenesis-of-sylvine-carnallite-polyhalite-and-cieserite-in-eskdale-borings-nos-1696717.webp)
The Paragenesis of Sylvine, Carnallite, Polyhalite, and ]Cieserite in Eskdale Borings Nos
667 The paragenesis of sylvine, carnallite, polyhalite, and ]cieserite in Eskdale borings nos. 3, 4, and 6, north-east Yorkshire. (With Plate XXV.) G. ARMSTRO~O, B.Se., F.G.S., K. C. DUNHAM, D.Sc., S.D., F.G.S., C. 0. HAI~VEY, B.Sc., A.R.C.S., F.R.I.C., P. A. SABINe, Ph.D., A.R.C.S., F.G.S., and W. F. WATERS, B.Sc., F.R.I.C. Geological Survey of Great Britain. [Read March 8, 1951.] INTRODUCTION. HE confirmation of the presence of potassium salts in the Permian T evaporites beneath the Whitby district, first found in the D'Arey Exploration Co.'s Eskdale no. 2 (Aislaby) boring (G. M. Lees and A. H. Taitt, 1945), and the discovery by Imperial Chemical Industries in their Eskdale no. 3 (Sleights), no. 4 (Sneaton), and no. 6 (Upgang) borings that these salts exist in workable quality and quantity, has been recorded in a recent historic paper by Dr. A. Fleck (1950). The successful outcome of this boring campaign may well lead to the outstanding development of the present century in economic mineralogy in Britain. In the course of the exploration the Geological Survey has examined in detail the cores obtained, and every facility has been granted by Imperial Chemical Industries for the removal of specimens from the cores for preservation in the national collection at the Geological Museum. The Petrographical Department of the Geological Survey has, on its side, provided reports on the mineralogy of the cores. The present brief summary of these reports, with some additional chemical, X-ray, and petrographical data, is published by permission of the Director of the Geological Survey and with the approval of Imperial Chemical Industries, Limited. -
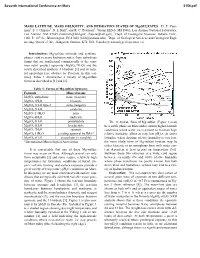
MARS LATITUDE, MARS OBLIQUITY, and HYDRATION STATES of Mg-SULFATES
Seventh International Conference on Mars 3156.pdf MARS LATITUDE, MARS OBLIQUITY, AND HYDRATION STATES OF Mg-SULFATES. D. T. Vani- man1, S. J. Chipera1, D. L Bish2, and R. C. Peterson3, 1Group EES-6, MS D462, Los Alamos National Laboratory, Los Alamos, NM 87545 ([email protected]; [email protected]), 2Dept. of Geological Sciences, Indiana Univ., 1001 E. 10th St., Bloomington, IN 47405 ([email protected]), 3Dept. of Geological Sciences and Geological Engi- neering, Queen’s Univ., Kingston, Ontario, K7L 3N6, Canada ([email protected]). Introduction: Mg-sulfate minerals and synthetic phases exist in many hydration states, from anhydrous forms that are synthesized commercially to the com- mon retail product epsomite (MgSO4·7H2O) and the newly described synthetic 11-hydrate [1] and its natu- ral equivalent [see abstract by Peterson in this vol- ume]. Table 1 summarizes a variety of Mg-sulfate forms as described in [1] and [2]. Table 1: Forms of Mg-sulfate hydrates Formula Mineral name MgSO4 anhydrous none (reagent) MgSO4·1H2O kieserite MgSO4·1H2O type 2 none (reagent) MgSO4·2H2O sanderite MgSO4·2.4H2O none MgSO ·4H O starkeyite 4 2 MgSO4·5H2O pentahydrite The 11-hydrate form of Mg-sulfate (Figure 1) may MgSO4·6H2O hexahydrite be a stable phase on Mars under current higher-latitude MgSO4·7H2O epsomite conditions where water ice is present to maintain high MgSO4·11H2O pending approval by IMA* relative humidity, albeit at very low pH2O. At lower MgSO4·nH2O amorphous (n variable) latitudes, where daytime relative humidity is very low, *International Mineralogical Association the more likely form of Mg-sulfate hydrate may be either kieserite or an amorphous form with water con- It is conceivable that any of these Mg-sulfate tent dependent at least in part on temperature [5,6]. -

Valles Marineris (VM) in a CRISM Observation Each Pixel of the Scene Has a Associated Spectrum
1 2 2 1 1 J. Flahaut *, C. Quantin ,P. Allemand , G.R. Davies and W. van Westrenen 1VU University Amsterdam, The Netherlands 2University Lyon 1, France. Although the first spacecrafts that flew over Mars showed the planet is rocky, cold, and sterile beneath A wide array of data (visible its hazy, pink sky, today's Martian imagery, spectroscopy, laser altimetry, wasteland hints at a formerly different gravity) from different missions are used in world where volcanoes once raged, order to identify, map and characterize geologic and flash floods rushed over the land [1]. Among our discoveries features and units at the surface of Mars. These about Mars, one stands out above all others: the possible presence data are collected, processed and gathered into a of liquid water on Mars, either in its ancient past or preserved in Geographic Information System (GIS) [2]. the subsurface today. Water is key because almost everywhere we find water on Earth, we find life. If Mars once had liquid Of particular interest are the data from the Compact water, or still does today, it is compelling to ask Reconnaissance Imaging Spectrometer for Mars (CRISM). whether any microscopic life forms could have CRISM is a hyperspectral imaging spectrometer onboard Mars developed on its surface. Reconnaissance Orbiter (NASA, 2005) which analyzed the light reflected by the planet’s surface thanks to 2 detectors in the visible/near infrared domains [3]. Absorptions in these domains are characteristic of the surface composition and especially the presence of hydrated minerals [4]. Crater counts (which are commmonly used in planetary science for relative dating of surfaces) performed in Valles Marineris (VM) In a CRISM observation each pixel of the scene has a associated spectrum. -

Physical Properties Data for Rock Salt QC100 .U556 V167;1981 C.2 NBS-PUB-C 1981
NATL INST OF STANDARDS & TECH R.I.C. NBS ,11100 161b7a PUBLICATIONS All 100989678 /Physical properties data for rock salt QC100 .U556 V167;1981 C.2 NBS-PUB-C 1981 NBS MONOGRAPH 167 U.S. DEPARTMENT OF COMMERCE / National Bureau of Standards NSRDS *f»fr nun^^ Physical Properties Data for Rock Salt NATIONAL BUREAU OF STANDARDS The National Bureau of Standards' was established by an act of Congress on March 3, 1901. The Bureau's overall goal is to strengthen and advance the Nation's science and technology and facilitate their effective application for public benefit. To this end, the Bureau conducts research and provides: (1) a basis for the Nation's physical measurement system, (2) scientific and technological services for industry and government, (3) a technical basis for equity in trade, and (4) technical services to promote public safety. The Bureau's technical work is per- formed by the National Measurement Laboratory, the National Engineering Laboratory, and the Institute for Computer Sciences and Technology. THE NATIONAL MEASUREMENT LABORATORY provides the national system of physical and chemical and materials measurement; coordinates the system with measurement systems of other nations and furnishes essential services leading to accurate and uniform physical and chemical measurement throughout the Nation's scientific community, industry, and commerce; conducts materials research leading to improved methods of measurement, standards, and data on the properties of materials needed by industry, commerce, educational institutions, and Government; provides advisory and research services to other Government agencies; develops, produces, and distributes Standard Reference Materials; and provides calibration services. The Laboratory consists of the following centers: Absolute Physical Quantities^ — Radiation Research — Thermodynamics and Molecular Science — Analytical Chemistry — Materials Science. -

MAGNESIUM SULFATE Chemical and Technical Assessment
MAGNESIUM SULFATE Chemical and Technical Assessment Revised by Madduri V. Rao, Ph.D. for the 68th JECFA (Original prepared by Yoko Kawamura, Ph.D. for the 63rd JECFA) 1. Summary Magnesium sulfate was evaluated by the Committee at its 63rd meeting. The outstanding information on functional uses other than nutrient supplement as well as information on commercial use of anhydrous magnesium sulfate and magnesium sulfate was requested for evaluation by the 68th JECFA meeting. Magnesium sulfate is commercially available as heptahydrate, monohydrate, anhydrous or dried form containing the equivalent of 2 - 3 waters of hydration. Magnesium sulfate occurs naturally in seawater, mineral springs and in minerals such as kieserite and epsomite. Magnesium sulfate heptahydrate is manufactured by dissolution of kieserite in water and subsequent crystallization of the heptahydrate. Magnesium sulfate is also prepared by sulfation of magnesium oxide. It is produced with one or seven molecules of water of hydration or in a dried form containing the equivalent of about 2 - 3 waters of hydration. Magnesium sulfate is available as brilliant colourless crystals, granular crystalline powder or white powder with a bitter salty cooling taste. Crystals effloresce in warm, dry air. It is freely soluble in water, very soluble in boiling water, and sparingly soluble in alcohol. Magnesium sulfate is used as a nutrient, firming agent and flavour enhancer. It is also used as a fermentation aid in the processing of beer and malt beverages. No food uses have been identified for the anhydrous form of magnesium sulfate. 2. Description Magnesium sulfate occurs with one (monohydrate), seven (heptahydrate), or no (anhydrous) molecules of water of hydration. -

Geology of the Tenth Potash Ore Zone, Permian Salado Formation
' OPEN FILE FEZOF2 146 GEOLOGY OF THE TENTH POTASH OREZONE: PERMIAN SALAD0 FORMATION, CARLSBAD DISTRICT, NEW MEXICO ROBERT C.M. GUNN AND JOHN M. HILLS I wish to thank the personnel of Noranda Mines Limited who allowed Gunn-to study and work on their potash exploration program, especially 0. Mr. Hinds, J. ,. Dr. J. J. M. Miller, Mr. J. Condon Mr.and J. F. Brewer., Information for this report could not have been obtained without their help and the friendly relation- ships with the following potash companies and individuals: Duval Corporation Mr. W. Blake Mr. M. P. Scroggin International Minerals and Chemical Corporation Mr. R. Koenig Kerr-McGee Chemical Corporation Mr. R. Lane National Potash Company Mr. P. Brewer United States Geological Survey Mr. C. L. Jones Independent Chemist who assayed Noranda potash core in Carlsbad Mr. T. J. Futch Independent Potash Consultant in Carlsbad Mr. E. H. Miller Dr. W. N. McAnulty, Dr. W. R. Roser and co-author Hills supervised Gun& original Masters thesis at Universityof Texas at El Paso upon which this paper is based. Dr. W. M. Schwerdtner of the University of Toronto has read the manuscript critically. Noranda Exploration Company has kindly permitted the publicationof insor- mation gathered from their properties. ABSTRACT The Tenth potash ore zone is a bedded evaporite deposit in the Permian (Ochoan) SaladoFormation in the Carlsbad district, Eddy and Lex counties, New Mexico.The rocksof the Tenth ore zone were precipitated in the super- saline part of the Permian basin and range from 6 to 10 feet (1.8 to 2.1 meters) .