Dosage-Sensitive Functions in Embryonic Development Drove the Survival of Genes on Sex-Specific Chromosomes in Snakes, Birds, and Mammals
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

P1382-Human Cellexp FLT-3 Ligand, Recombinant Mouse
FOR RESEARCH ONLY! Human CellExp™ FLT-3 Ligand, Mouse Recombinant CATALOG #: P1382-50 P1382-10 AMOUNT: 50 µg 10 µg FLT3LG, FL, FLT3L, Flt3 ligand ALTERNATE NAMES: MOL. WT. The protein has a calculated MW of 20.2 kDa. The protein migrates as 25-33 kDa under reducing (R) condition (SDS-PAGE) due to glycosylation. SOURCE: HEK 293 cells PURITY: >90% as determined by SDS-PAGE. ENDOTOXIN: Less than 1.0 EU per μg by the LAL method FORM: Lyophilized FORMULATION: Lyophilized from 0.22 μm filtered solution in PBS, pH7.4. Normally trehalose is added as protectant before lyophilization. Centrifuge the vial prior to opening. Reconstitute in sterile deionized water to a concentration of 100 µg/ml. RECONSTITUTION: Do not vortex. It is recommended to store at -20°C. SPECIFIC ACTIVITY: Immobilized Mouse Flt-3 Ligand, His Tag at 1 μg/mL (100 μL/well) can bind Human Flt-3/Flk-2 Fc Chimera with a linear range of 1-5 ng/mL STORAGE CONDITIONS: Store at -20°C. After reconstitution, aliquot and store at -20°C and use within 3 months. Avoid repeated freezing and thawing cycles. DESCRIPTION: FMS-like tyrosine kinase 3 ligand (Flt-3 Ligand) is also known as FL, Flt3L and FLT3LG, is an α-helical cytokine that promotes the differentiation of multiple hematopoietic cell lineages. FLT3LG is expressed as a noncovalentlylinked dimer by T cells and bone marrow and thymic fibroblasts. Each 36 kDa chain carries approximately 12 kDa of N- and O- linked carbohydrates. FLT3LG is structurally homologous to stem cell factor (SCF) and colony stimulating facor 1 (CSF-1). -

Flt3 Ligand (FLT3LG) (NM 001278637) Human Tagged ORF Clone Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RG236103 Flt3 ligand (FLT3LG) (NM_001278637) Human Tagged ORF Clone Product data: Product Type: Expression Plasmids Product Name: Flt3 ligand (FLT3LG) (NM_001278637) Human Tagged ORF Clone Tag: TurboGFP Symbol: FLT3LG Synonyms: FL; FLG3L; FLT3L Vector: pCMV6-AC-GFP (PS100010) E. coli Selection: Ampicillin (100 ug/mL) Cell Selection: Neomycin ORF Nucleotide >RG236103 representing NM_001278637. Sequence: Blue=ORF Red=Cloning site Green=Tag(s) GCTCGTTTAGTGAACCGTCAGAATTTTGTAATACGACTCACTATAGGGCGGCCGGGAATTCGTCGACTG GATCCGGTACCGAGGAGATCTGCCGCCGCGATCGCC ATGGAGCGGCTCAAGACTGTCGCTGGGTCCAAGATGCAAGGCTTGCTGGAGCGCGTGAACACGGAGATA CACTTTGTCACCAAATGTGCCTTTCAGCCCCCCCCCAGCTGTCTTCGCTTCGTCCAGACCAACATCTCC CGCCTCCTGCAGGAGACCTCCGAGCAGCTGGTGGCGCTGAAGCCCTGGATCACTCGCCAGAACTTCTCC CGGTGCCTGGAGCTGCAGTGTCAGCCCGACTCCTCAACCCTGCCACCCCCATGGAGTCCCCGGCCCCTG GAGGCCACAGCCCCGACAGCCCCGCAGCCCCCTCTGCTCCTCCTACTGCTGCTGCCCGTGGGCCTCCTG CTGCTGGCCGCTGCCTGGTGCCTGCACTGGCAGAGGACGCGGCGGAGGACACCCCGCCCTGGGGAGCAG GTGCCCCCCGTCCCCAGTCCCCAGGACCTGCTGCTTGTGGAGCAC ACGCGTACGCGGCCGCTCGAG - GFP Tag - GTTTAAAC Protein Sequence: >Peptide sequence encoded by RG236103 Blue=ORF Red=Cloning site Green=Tag(s) MERLKTVAGSKMQGLLERVNTEIHFVTKCAFQPPPSCLRFVQTNISRLLQETSEQLVALKPWITRQNFS RCLELQCQPDSSTLPPPWSPRPLEATAPTAPQPPLLLLLLLPVGLLLLAAAWCLHWQRTRRRTPRPGEQ VPPVPSPQDLLLVEH TRTRPLEMESDESGLPAMEIECRITGTLNGVEFELVGGGEGTPEQGRMTNKMKSTKGALTFSPYLLSHV -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -

Data Appendix to the White Paper: Multiplex Analysis of Inflammatory Proteins: a Comparative Study Across Multiple Platforms
Data appendix to the White paper: Multiplex analysis of inflammatory proteins: A comparative study across multiple platforms Appendix content This appendix contains several graphs. Olink results are shown in the left-hand column, MSD in the middle and Luminex to the right. Please note that the y-axis scales differ between platforms for some assays. In this study, Olink, MSD and Luminex had an overlap of 20 assays. Olink and MSD had an additional overlap of 13 assays, and those results are therefore presented without associated Luminex data. The MSD V-plex platform was used for as many assays as possible. U-plex was used for the following assays: FLT3LG, G-CSF, IL-17F, IL-33, I-TAC, MCP-2, MIP-3b, SDF-1a and TRAIL. For these assays LLOQ and ULOQ is not available. Dilution The first section shows parallelism between Olink, MSD and Luminex, calculated using dilution series in buffer. Each sample is displayed with the same color in the plot throughout all charts. Visually the dilution series should be linear. Note that Luminex displays higher concentrations and changed order of samples compared to Olink and MSD for several assays. Concentrations below the dotted line are under the Lower limit of quantification (LLOQ). Note that nine assays were measured on an MSD U-PLEX panel and has no LLOQ or ULOQ limit calculated. Linearity The second section shows platform linearity. The graphs display measured and theoretical concentrations. Bars indicate standard deviation and the percentage represents accuracy for each point. Validation data The third section includes a table that contains validation data for Olink Explore 1536 and Olink Target 48 Cytokine . -

Anti-CD135 / FLT3 Antibody (ARG43119)
Product datasheet [email protected] ARG43119 Package: 50 μg anti-CD135 / FLT3 antibody Store at: -20°C Summary Product Description Rabbit Polyclonal antibody recognizes CD135 / FLT3 Tested Reactivity Hu Tested Application IHC-P, WB Host Rabbit Clonality Polyclonal Isotype IgG Target Name CD135 / FLT3 Antigen Species Human Immunogen Recombinant protein corresponding to E61-E294 of Human CD135 / FLT3. Conjugation Un-conjugated Alternate Names CD135; FLK2; Receptor-type tyrosine-protein kinase FLT3; FLK-2; STK-1; STK1; FL cytokine receptor; FLT-3; Stem cell tyrosine kinase 1; Fetal liver kinase-2; Fms-like tyrosine kinase 3; CD antigen CD135; EC 2.7.10.1 Application Instructions Application table Application Dilution IHC-P 1:200 - 1:1000 WB 1:500 - 1:2000 Application Note IHC-P: Antigen Retrieval: Heat mediation was performed in Citrate buffer (pH 6.0) for 20 min. * The dilutions indicate recommended starting dilutions and the optimal dilutions or concentrations should be determined by the scientist. Calculated Mw 113 kDa Observed Size ~ 130 kDa Properties Form Liquid Purification Immunogen affinity purified. Buffer 0.2% Na2HPO4, 0.9% NaCl, 0.05% Sodium azide and 4% Trehalose. Preservative 0.05% Sodium azide Stabilizer 4% Trehalose Concentration 0.5 mg/ml Storage instruction For continuous use, store undiluted antibody at 2-8°C for up to a week. For long-term storage, aliquot and store at -20°C or below. Storage in frost free freezers is not recommended. Avoid repeated www.arigobio.com 1/3 freeze/thaw cycles. Suggest spin the vial prior to opening. The antibody solution should be gently mixed before use. -

Supplementary Figure 1. Network Map Associated with Upregulated Canonical Pathways Shows Interferon Alpha As a Key Regulator
Supplementary Figure 1. Network map associated with upregulated canonical pathways shows interferon alpha as a key regulator. IPA core analysis determined interferon-alpha as an upstream regulator in the significantly upregulated genes from RNAseq data from nasopharyngeal swabs of COVID-19 patients (GSE152075). Network map was generated in IPA, overlaid with the Coronavirus Replication Pathway. Supplementary Figure 2. Network map associated with Cell Cycle, Cellular Assembly and Organization, DNA Replication, Recombination, and Repair shows relationships among significant canonical pathways. Significant pathways were identified from pathway analysis of RNAseq from PBMCs of COVID-19 patients. Coronavirus Pathogenesis Pathway was also overlaid on the network map. The orange and blue colors in indicate predicted activation or predicted inhibition, respectively. Supplementary Figure 3. Significant biological processes affected in brochoalveolar lung fluid of severe COVID-19 patients. Network map was generated by IPA core analysis of differentially expressed genes for severe vs mild COVID-19 patients in bronchoalveolar lung fluid (BALF) from scRNA-seq profile of GSE145926. Orange color represents predicted activation. Red boxes highlight important cytokines involved. Supplementary Figure 4. 10X Genomics Human Immunology Panel filtered differentially expressed genes in each immune subset (NK cells, T cells, B cells, and Macrophages) of severe versus mild COVID-19 patients. Three genes (HLA-DQA2, IFIT1, and MX1) were found significantly and consistently differentially expressed. Gene expression is shown per the disease severity (mild, severe, recovered) is shown on the top row and expression across immune cell subsets are shown on the bottom row. Supplementary Figure 5. Network map shows interactions between differentially expressed genes in severe versus mild COVID-19 patients. -

CD135 / FLT3 Antibody (Internal) Rabbit Polyclonal Antibody Catalog # ALS15658
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 CD135 / FLT3 Antibody (Internal) Rabbit Polyclonal Antibody Catalog # ALS15658 Specification CD135 / FLT3 Antibody (Internal) - Product Information Application IF Primary Accession P36888 Reactivity Human Host Rabbit Clonality Polyclonal Calculated MW 113kDa KDa CD135 / FLT3 Antibody (Internal) - Additional Information Gene ID 2322 Other Names Receptor-type tyrosine-protein kinase FLT3, Immunofluorescence of FLT3 in human 2.7.10.1, FL cytokine receptor, Fetal liver kidney tissue with FLT3 antibody at 20 ug/ml. kinase-2, FLK-2, Fms-like tyrosine kinase 3, FLT-3, Stem cell tyrosine kinase 1, STK-1, CD135, FLT3, CD135, FLK2, STK1 CD135 / FLT3 Antibody (Internal) - Background Target/Specificity Human FLT3 / CD135. At least three Tyrosine-protein kinase that acts as isoforms of FLT3 are known to exist; this cell-surface receptor for the cytokine FLT3LG antibody will detect all isoforms. and regulates differentiation, proliferation and survival of hematopoietic progenitor cells and Reconstitution & Storage of dendritic cells. Promotes phosphorylation of Long term: -20°C; Short term: +4°C. Avoid SHC1 and AKT1, and activation of the repeat freeze-thaw cycles. downstream effector MTOR. Promotes activation of RAS signaling and Precautions phosphorylation of downstream kinases, CD135 / FLT3 Antibody (Internal) is for including MAPK1/ERK2 and/or MAPK3/ERK1. research use only and not for use in Promotes phosphorylation of FES, FER, diagnostic or therapeutic procedures. PTPN6/SHP, PTPN11/SHP-2, PLCG1, and STAT5A and/or STAT5B. Activation of wild-type FLT3 causes only marginal activation of CD135 / FLT3 Antibody (Internal) - Protein STAT5A or STAT5B. -

KITLG & FLT3LG Protein Protein Interaction Antibody Pair
KITLG & FLT3LG Protein Protein Interaction Antibody Pair Catalog # : DI0207 規格 : [ 1 Set ] List All Specification Application Image Product This protein protein interaction antibody pair set comes with two In situ Proximity Ligation Assay (Cell) Description: antibodies to detect the protein-protein interaction, one against the KITLG protein, and the other against the FLT3LG protein for use in in situ Proximity Ligation Assay. See Publication Reference below. Reactivity: Human Quality Control Protein protein interaction immunofluorescence result. Testing: Representative image of Proximity Ligation Assay of protein-protein interactions between KITLG and FLT3LG. HeLa cells were stained with anti-KITLG rabbit purified polyclonal antibody 1:1200 and anti-FLT3LG mouse monoclonal antibody 1:50. Each red dot represents the detection of protein-protein interaction complex. The images were analyzed using an optimized freeware (BlobFinder) download from The Centre for Image Analysis at Uppsala University. Supplied Antibody pair set content: Product: 1. KITLG rabbit purified polyclonal antibody (20 ug) 2. FLT3LG mouse monoclonal antibody (40 ug) *Reagents are sufficient for at least 30-50 assays using recommended protocols. Storage Store reagents of the antibody pair set at -20°C or lower. Please aliquot Instruction: to avoid repeated freeze thaw cycle. Reagents should be returned to - Page 1 of 3 2016/5/19 20°C storage immediately after use. MSDS: Download Publication Reference 1. An analysis of protein-protein interactions in cross-talk pathways reveals CRKL as a novel prognostic marker in hepatocellular carcinoma. Liu CH, Chen TC, Chau GY, Jan YH, Chen CH, Hsu CN, Lin KT, Juang YL, Lu PJ, Cheng HC, Chen MH, Chang CF, Ting YS, Kao CY, Hsiao M, Huang CY. -
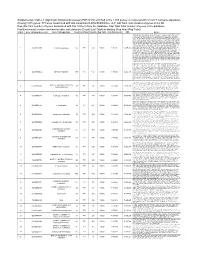
(FDR<0.05) Enriched in the 1015 Genes Co-Clustered with Known T
Supplementary Table 3. Significant biological processes (FDR<0.05) enriched in the 1,015 genes co-clustered with known T cell gene signatures. Among 1,015 genes, 771 were associated with GO annotation in DAVID database v6.7. List Total: total number of genes in my list. Pop Hits: total number of genes associated with this GO term from the database. Pop Total: total number of genes in the database. Fold Enrichment: relative enrichment ratio, calculated by (Count)/(List Total) divided by (Pop Hits)/(Pop Total). Index Gene Ontology Accession Gene Ontology Name Count List Total Pop Hits Pop Total Fold Enrichment FDR Genes AQP9, C1QC, B2M, LILRA1, LILRA2, CLEC4E, S1PR4, LILRA4, IFNG, LILRA6, CLEC4A, VNN1, ERAP2, FAS, CRTAM, C5AR1, GBP5, NCF2, NCF1, NCF4, SERPING1, HLA-DQA2, HLA-DQA1, PDCD1LG2, LILRB1, CCR9, C1QA, C1QB, LILRB2, CCR7, CCR6, UNC13D, CCR5, CD40LG, CCR4, LILRB3, CCR2, LILRB4, HLA-DPA1, VSIG4, HLA-DRA, IL1R2, IL1R1, HLA-DRB1, OAS3, ACP5, OAS1, OAS2, CD74, IFI35, ZAP70, FCER1G, HLA-DRB5, HLA-DPB1, HLA-DOA, HLA-DOB, DHX58, BLNK, IL23R, KIR2DS4, CD300C, SLAMF7, OASL, RGS1, APOL1, CD300A, HMHB1, CD209, CLEC7A, LY86, LY9, CLNK, FCRL4, SH2D1A, NOD2, HAMP, CCL3L1, CCL3L3, TICAM2, ICAM1, GZMA, CMKLR1, LY96, WAS, IL18BP, LAX1, TNFSF12- TNFSF13, HLA-DQB1, CSF2, GPR183, CCR1, GPR65, CXCL9, NCF1C, IL7R, CLEC10A, CCL24, CCL22, CYP27B1, CCL23, FCGR1C, FTHL3, FCGR1A, FCGR1B, BCL3, C2, CD27, CD28, FYB, IL18R1, IL7, CD1C, CTLA4, CCL19, CD1B, CD1A, TRIM22, CD180, CD1E, CCL18, CCL17, CCL13, FCGR2B, FCGR2C, P2RY14, LIME1, CD14, IL16, IL18, TLR1, TNFSF15, -
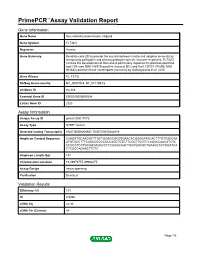
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name fms-related tyrosine kinase 3 ligand Gene Symbol FLT3LG Organism Human Gene Summary Dendritic cells (DCs) provide the key link between innate and adaptive immunity by recognizing pathogens and priming pathogen-specific immune responses. FLT3LG controls the development of DCs and is particularly important for plasmacytoid DCs and CD8 (see MIM 186910)-positive classical DCs and their CD103 (ITGAE; MIM 604682)-positive tissue counterparts (summary by Sathaliyawala et al. 2010 Gene Aliases FL, FLT3L RefSeq Accession No. NC_000019.9, NT_011109.16 UniGene ID Hs.428 Ensembl Gene ID ENSG00000090554 Entrez Gene ID 2323 Assay Information Unique Assay ID qHsaCID0017073 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000204637, ENST00000344019 Amplicon Context Sequence CAAGATGCAAGGCTTGCTGGAGCGCGTGAACACGGAGATACACTTTGTCACCAA ATGTGCCTTTCAGCCCCCCCCCAGCTGTCTTCGCTTCGTCCAGACCAACATCTC CCGCCTCCTGCAGGAGACCTCCGAGCAGCTGGTGGCGCTGAAGCCCTGGATCA CTCGCCAGAACTTCTC Amplicon Length (bp) 147 Chromosome Location 19:49979757-49982275 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 101 R2 0.9996 cDNA Cq 24.33 cDNA Tm (Celsius) 88 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq 35.08 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report FLT3LG, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above -

(FLT3LG) (NM 001459) Human Recombinant Protein Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TP322242 Flt3 ligand (FLT3LG) (NM_001459) Human Recombinant Protein Product data: Product Type: Recombinant Proteins Description: Recombinant protein of human fms-related tyrosine kinase 3 ligand (FLT3LG) Species: Human Expression Host: HEK293T Tag: C-Myc/DDK Predicted MW: 26.2 kDa Concentration: >50 ug/mL as determined by microplate BCA method Purity: > 80% as determined by SDS-PAGE and Coomassie blue staining Buffer: 25 mM Tris.HCl, pH 7.3, 100 mM glycine, 10% glycerol Bioactivity: FLT-3LG generate dendritic cell differentiation: Fms-like tyrosine kinase 3 ligand (Flt3L) is a nonredundant cytokine required for dendritic cell (DC) homeostasis in lymphoid tissues. FLT3L is commonly used to generate pDCs in vitro.In this experiment, 300,000 unfractioned mouse bone marrow cells were cultured with purified recombinant human FLT-3L (OriGene, TP322242) at indicated concentrations. Total live cells were counted at the day 8. Preparation: Recombinant protein was captured through anti-DDK affinity column followed by conventional chromatography steps. Storage: Store at -80°C. Stability: Stable for 12 months from the date of receipt of the product under proper storage and handling conditions. Avoid repeated freeze-thaw cycles. RefSeq: NP_001450 Locus ID: 2323 UniProt ID: P49771 RefSeq Size: 1074 Cytogenetics: 19q13.33 RefSeq ORF: 705 Synonyms: FL; FLG3L; FLT3L This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 Flt3 ligand (FLT3LG) (NM_001459) Human Recombinant Protein – TP322242 Summary: Dendritic cells (DCs) provide the key link between innate and adaptive immunity by recognizing pathogens and priming pathogen-specific immune responses. -

Flt3 Polyclonal Antibody Catalog # AP69913
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 Flt3 Polyclonal Antibody Catalog # AP69913 Specification Flt3 Polyclonal Antibody - Product Information Application WB Primary Accession P36888 Reactivity Human, Mouse Host Rabbit Clonality Polyclonal Flt3 Polyclonal Antibody - Additional Information Gene ID 2322 Other Names FLT3; CD135; FLK2; STK1; Receptor-type tyrosine-protein kinase FLT3; FL cytokine receptor; Fetal liver kinase-2; FLK-2; Fms-like tyrosine kinase 3; FLT-3; Stem cell tyrosine kinase 1; STK-1; CD antigen CD135 Dilution WB~~Western Blot: 1/500 - 1/2000. ELISA: Flt3 Polyclonal Antibody - Background 1/20000. Not yet tested in other applications. Tyrosine-protein kinase that acts as cell-surface receptor for the cytokine FLT3LG Format and regulates differentiation, proliferation and Liquid in PBS containing 50% glycerol, 0.5% survival of hematopoietic progenitor cells and BSA and 0.02% sodium azide. of dendritic cells. Promotes phosphorylation of SHC1 and AKT1, and activation of the Storage Conditions downstream effector MTOR. Promotes -20℃ activation of RAS signaling and phosphorylation of downstream kinases, including MAPK1/ERK2 and/or MAPK3/ERK1. Flt3 Polyclonal Antibody - Protein Information Promotes phosphorylation of FES, FER, PTPN6/SHP, PTPN11/SHP-2, PLCG1, and Name FLT3 STAT5A and/or STAT5B. Activation of wild-type FLT3 causes only marginal activation of Synonyms CD135, FLK2, STK1 STAT5A or STAT5B. Mutations that cause constitutive kinase activity promote cell Function proliferation and resistance to apoptosis via Tyrosine-protein kinase that acts as the activation of multiple signaling pathways. cell-surface receptor for the cytokine FLT3LG and regulates differentiation, proliferation and survival of hematopoietic progenitor cells and of dendritic cells.