Chemistry with Graphene and Graphene Oxide - Challenges for Synthetic Chemists Siegfried Eigler* and Andreas Hirsch*
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Graphite Materials for the U.S
ANRIC your success is our goal SUBSECTION HH, Subpart A Timothy Burchell CNSC Contract No: 87055-17-0380 R688.1 Technical Seminar on Application of ASME Section III to New Materials for High Temperature Reactors Delivered March 27-28, 2018, Ottawa, Canada TIM BURCHELL BIOGRAPHICAL INFORMATION Dr. Tim Burchell is Distinguished R&D staff member and Team Lead for Nuclear Graphite in the Nuclear Material Science and Technology Group within the Materials Science and Technology Division of the Oak Ridge National Laboratory (ORNL). He is engaged in the development and characterization of carbon and graphite materials for the U.S. Department of Energy. He was the Carbon Materials Technology (CMT) Group Leader and was manager of the Modular High Temperature Gas-Cooled Reactor Graphite Program responsible for the research project to acquire reactor graphite property design data. Currently, Dr. Burchell is the leader of the Next Generation Nuclear Plant graphite development tasks at ORNL. His current research interests include: fracture behavior and modeling of nuclear-grade graphite; the effects of neutron damage on the structure and properties of fission and fusion reactor relevant carbon materials, including isotropic and near isotropic graphite and carbon-carbon composites; radiation creep of graphites, the thermal physical properties of carbon materials. As a Research Officer at Berkeley Nuclear Laboratories in the U.K. he monitored the condition of graphite moderators in gas-cooled power reactors. He is a Battelle Distinguished Inventor; received the Hsun Lee Lecture Award from the Chinese Academy of Science’s Institute of Metals Research in 2006 and the ASTM D02 Committee Eagle your success is our goal Award in 2015. -

The Era of Carbon Allotropes Andreas Hirsch
commentary The era of carbon allotropes Andreas Hirsch Twenty-five years on from the discovery of 60C , the outstanding properties and potential applications of the synthetic carbon allotropes — fullerenes, nanotubes and graphene — overwhelmingly illustrate their unique scientific and technological importance. arbon is the element in the periodic consist of extended networks of sp3- and 1985, with the advent of fullerenes (Fig. 1), table that provides the basis for life sp2 -hybridized carbon atoms, respectively. which were observed for the first time by Con Earth. It is also important for Both forms show unique physical properties Kroto et al.3. This serendipitous discovery many technological applications, ranging such as hardness, thermal conductivity, marked the beginning of an era of synthetic from drugs to synthetic materials. This lubrication behaviour or electrical carbon allotropes. Now, as we celebrate role is a consequence of carbon’s ability conductivity. Conceptually, many other buckminsterfullerene’s 25th birthday, it is to bind to itself and to nearly all elements ways to construct carbon allotropes are also the time to reflect on a growing family in almost limitless variety. The resulting possible by altering the periodic binding of synthetic carbon allotropes, which structural diversity of organic compounds motif in networks consisting of sp3-, sp2- includes the synthesis of carbon nanotubes and molecules is accompanied by a broad and sp-hybridized carbon atoms1,2. As a in 19914 and the rediscovery of graphene range of -

Reduction Kinetics of Graphene Oxide Determined by Electrical Transport Measurements and Temperature Programmed Desorption
J. Phys. Chem. C XXXX, xxx, 000 A Reduction Kinetics of Graphene Oxide Determined by Electrical Transport Measurements and Temperature Programmed Desorption Inhwa Jung,† Daniel A. Field,‡ Nicholas J. Clark,‡ Yanwu Zhu,† Dongxing Yang,† Richard D. Piner,† Sasha Stankovich,§ Dmitriy A. Dikin,§ Heike Geisler,| Carl A. Ventrice, Jr.,*,‡ and Rodney S. Ruoff*,† Department of Mechanical Engineering and the Texas Materials Institute, UniVersity of Texas at Austin, Austin, Texas, 78712, Department of Physics, Texas State UniVersity, San Marcos, Texas, 78666, Department of Mechanical Engineering, Northwestern UniVersity, EVanston, Illinois, 60208, and Insitute for EnVironmental and Industrial Science, Texas State UniVersity, San Marcos, Texas, 78666 ReceiVed: May 11, 2009; ReVised Manuscript ReceiVed: August 31, 2009 The thermal stability and reduction kinetics of graphene oxide were studied by measuring the electrical resistivity of single-layer graphene films at various stages of reduction in high vacuum and by performing temperature programmed desorption (TPD) measurements of multilayer films in ultrahigh vacuum. The graphene oxide was exfoliated from the graphite oxide source material by slow-stirring in aqueous solution, which produces single-layer platelets with an average lateral size of ∼10 µm. From the TPD measurements, it was determined that the primary desorption products of the graphene oxide films for temperatures up to 300 °C are H2O, CO2, and CO, with only trace amounts of O2 being detected. Resistivity measurements on individual single-layer graphene oxide platelets resulted in an activation energy of 37 ( 1 kcal/mol. The TPD measurements of multilayer films of graphene oxide platelets give an activation energy of 32 ( 4 kcal/mol. 1. -

Coal Characteristics
CCTR Indiana Center for Coal Technology Research COAL CHARACTERISTICS CCTR Basic Facts File # 8 Brian H. Bowen, Marty W. Irwin The Energy Center at Discovery Park Purdue University CCTR, Potter Center, 500 Central Drive West Lafayette, IN 47907-2022 http://www.purdue.edu/dp/energy/CCTR/ Email: [email protected] October 2008 1 Indiana Center for Coal Technology Research CCTR COAL FORMATION As geological processes apply pressure to peat over time, it is transformed successively into different types of coal Source: Kentucky Geological Survey http://images.google.com/imgres?imgurl=http://www.uky.edu/KGS/coal/images/peatcoal.gif&imgrefurl=http://www.uky.edu/KGS/coal/coalform.htm&h=354&w=579&sz= 20&hl=en&start=5&um=1&tbnid=NavOy9_5HD07pM:&tbnh=82&tbnw=134&prev=/images%3Fq%3Dcoal%2Bphotos%26svnum%3D10%26um%3D1%26hl%3Den%26sa%3DX 2 Indiana Center for Coal Technology Research CCTR COAL ANALYSIS Elemental analysis of coal gives empirical formulas such as: C137H97O9NS for Bituminous Coal C240H90O4NS for high-grade Anthracite Coal is divided into 4 ranks: (1) Anthracite (2) Bituminous (3) Sub-bituminous (4) Lignite Source: http://cc.msnscache.com/cache.aspx?q=4929705428518&lang=en-US&mkt=en-US&FORM=CVRE8 3 Indiana Center for Coal Technology Research CCTR BITUMINOUS COAL Bituminous Coal: Great pressure results in the creation of bituminous, or “soft” coal. This is the type most commonly used for electric power generation in the U.S. It has a higher heating value than either lignite or sub-bituminous, but less than that of anthracite. Bituminous coal -

Static and Dynamic Behavior of Polymer/Graphite Oxide Nanocomposites Before and After Thermal Reduction
polymers Article Static and Dynamic Behavior of Polymer/Graphite Oxide Nanocomposites before and after Thermal Reduction Kiriaki Chrissopoulou 1,* , Krystalenia Androulaki 1,2, Massimiliano Labardi 3 and Spiros H. Anastasiadis 1,2 1 Institute of Electronic Structure and Laser, Foundation for Research and Technology-Hellas, N. Plastira 100, 700 13 Heraklion Crete, Greece; [email protected] (K.A.); [email protected] (S.H.A.) 2 Department of Chemistry, University of Crete, P.O. Box 2208, 710 03 Heraklion Crete, Greece 3 CNR-IPCF, c/o Physics Department, University of Pisa, Largo Pontecorvo 3, 56127 Pisa, Italy; [email protected] * Correspondence: [email protected]; Tel.: +30-281-039-1255 Abstract: Nanocomposites of hyperbranched polymers with graphitic materials are investigated with respect to their structure and thermal properties as well as the dynamics of the polymer probing the effect of the different intercalated or exfoliated structure. Three generations of hyperbranched polyester polyols are mixed with graphite oxide (GO) and the favorable interactions between the polymers and the solid surfaces lead to intercalated structure. The thermal transitions of the confined chains are suppressed, whereas their dynamics show similarities and differences with the dynamics of the neat polymers. The three relaxation processes observed for the neat polymers are observed in the nanohybrids as well, but with different temperature dependencies. Thermal reduction of the graphite oxide in the presence of the polymer to produce reduced graphite oxide (rGO) reveals an increase in the reduction temperature, which is accompanied by decreased thermal stability of the polymer. Citation: Chrissopoulou, K.; The de-oxygenation of the graphite oxide leads to the destruction of the intercalated structure and Androulaki, K.; Labardi, M.; to the dispersion of the rGO layers within the polymeric matrix because of the modification of the Anastasiadis, S.H. -

Properties and Characteristics of Graphite
SPECIALTY MATERIALS PROPERTIES AND CHARACTERISTICS OF GRAPHITE For the semiconductor industry May 2013 INTRODUCTION Introduction Table of Contents As Entegris has continued to grow its market share of Introduction .............................................................1 manufactured graphite material and to expand their usage into increasingly complex areas, the need to Structure ..................................................................2 be more technically versed in both Entegris’ POCO® graphite and other manufactured graphite properties Apparent Density .....................................................6 and how they are tested has also grown. It is with this Porosity ....................................................................8 thought in mind that this primer on properties and characteristics of graphite was developed. Hardness ................................................................11 POCO, now owned by Entegris, has been a supplier Compressive Strength .............................................12 to the semiconductor industry for more than 35 years. Products include APCVD wafer carriers, E-Beam cruci- Flexural Strength ...................................................14 bles, heaters (small and large), ion implanter parts, Tensile Strength .....................................................16 LTO injector tubes, MOCVD susceptors, PECVD wafer trays and disk boats, plasma etch electrodes, quartz Modulus of Elasticity .............................................18 replacement parts, sealing -

Synthesis of Graphene.Pdf
Materials Chemistry and Physics 206 (2018) 7e11 Contents lists available at ScienceDirect Materials Chemistry and Physics journal homepage: www.elsevier.com/locate/matchemphys Synthesis of graphene via ultra-sonic exfoliation of graphite oxide and its electrochemical characterization Hassnain Asgar a, K.M. Deen b, Usman Riaz a, Zia Ur Rahman c, Umair Hussain Shah a, * Waseem Haider a, c, a School of Engineering and Technology, Central Michigan University, Mt. Pleasant, MI 48859, USA b Department of Materials Engineering, University of British Columbia, Vancouver, BC V6T 1Z4, Canada c Science of Advanced Materials, Central Michigan University, Mt. Pleasant, MI 48859, USA highlights A relatively direct synthesis method for production of graphene is presented. IR, Raman and XRD analyses confirmed formation of graphene starting from graphite. XPS and TEM characterization validated the formation of graphene. Electrochemical response of GO and graphene was evaluated in de-aerated 0.5M KOH. Presence of functional groups in GO resulted in improved values of Rct and Ceff,p. abstract A direct method of producing graphene from graphite oxide (GO) via ultra-sonication is presented in this work. The synthesis of graphene was validated through IR, XRD, Raman, and XPS analyses. Moreover, the diffraction pattern obtained from TEM also validated the formation of graphene with char- acteristics (002) plane. The electrochemical behavior of GO and graphene was evaluated by electrochemical impedance spectroscopy and linear sweep voltammetry in 0.5M KOH solution. The relatively larger effective pseudocapacitance and broad current peak exhibited by GO in the LSV plots was related with the dominant adsorption of ‘Hads’ during reduction of water. -

Graphene: a Peculiar Allotrope of Carbon
Graphene: A Peculiar Allotrope Of Carbon Laxmi Nath Bhattarai Department of Physics, Butwal Multiple Campus Correspondence: [email protected] Abstract Graphene is a two dimensional one atom thick allotrope of Carbon. Electrons in grapheme behave as massless relativistic particles. It is a 2 dimensional nanomaterial with many peculiar properties. In grapheme both integral and fractional quantum Hall effects are observed. Many practical application are seen from use of Graphene material. Keywords: Graphene, allotrope, Quantum Hall effect, nanomaterial. Introduction Graphene is a two dimensional allotropic form of Honey comb structure of Graphene carbon. This was discovered in 2004. Diamond and Graphene is considered as the mother of all graphitic Graphite are three dimensional allotropes of carbon materials because it is the building block of carbon which are known from ancient time. One dimensional materials of all other dimensions (Srinivasan, 2007). carbon nanotubes were discovered in 1991 and Griphite is obtained by the stacking of Graphene zero dimensional Fullerenes were discovered in layers, Diamond can be obtained from Graphene under 1985. Graphene was experimentally extracted from extreme pressure and temperatures. One-dimensional 3-dimensional Graphite by Physisists Andre Geim and carbon nanotubes can be obtained by rolling and Konstantin Novoselov of Manchester University UK Zero–dimensional Fullerenes is obtained by wrapping in 2004. For their remarkable work they were awarded Graphene layers. the Nobel Prize of Physics for the year 2010. Properties Structure The young material Graphene is found to have Graphene is a mono layer of Carbon atoms packed following unique properties: into a honeycomb crystal structure. Graphene sheets 2 are one atom thick 2-dimensional layers of sp – bonded a) It is the thinnest material in the universe and the carbon. -

The Preparation of Graphene Oxide and Its Derivatives and Their Application in Bio-Tribological Systems
Lubricants 2014, 2, 137-161; doi:10.3390/lubricants2030137 OPEN ACCESS lubricants ISSN 2075-4442 www.mdpi.com/journal/lubricants Review The Preparation of Graphene Oxide and Its Derivatives and Their Application in Bio-Tribological Systems Jianchang Li 1,2, Xiangqiong Zeng 1,*, Tianhui Ren 2 and Emile van der Heide 1,3 1 Laboratory for Surface Technology and Tribology, University of Twente, P.O. Box 217, 7500 AE Enschede, The Netherlands; E-Mails: [email protected] (J.L.); [email protected] (E.H.) 2 School of Chemistry and Chemical Engineering, Key Laboratory for Thin Film and Microfabrication of the Ministry of Education, Shanghai Jiao Tong University, Shanghai 200240, China; E-Mail: [email protected] 3 Netherlands Organisation for Applied Scientific Research—TNO, P.O. Box 6235, NL-5600 HE Eindhoven, The Netherlands * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +31-53-489-4390. Received: 20 June 2014; in revised form: 15 August 2014 / Accepted: 22 August 2014 / Published: 24 September 2014 Abstract: Graphene oxide (GO) can be readily modified for particular applications due to the existence of abundant oxygen-containing functional groups. Graphene oxide-based materials (GOBMs), which are biocompatible and hydrophilic, have wide potential applications in biomedical engineering and biotechnology. In this review, the preparation and characterization of GO and its derivatives are discussed at first. Subsequently, the biocompatibility and tribological behavior of GOBMs are reviewed. Finally, the applications of GOBMs as lubricants in bio-tribological systems are discussed in detail. Keywords: graphene oxide; graphene oxide-based materials; biocompatibility; bio-tribology 1. -

Studies of Reduced Graphene Oxide and Graphite Oxide in the Aspect of Their Possible Application in Gas Sensors
sensors Article Studies of Reduced Graphene Oxide and Graphite Oxide in the Aspect of Their Possible Application in Gas Sensors Sabina Drewniak 1,*, Roksana Muzyka 2, Agnieszka Stolarczyk 3, Tadeusz Pustelny 1, Michalina Kotyczka-Mora ´nska 2 and Maciej Setkiewicz 1 Received: 25 November 2015; Accepted: 8 January 2016; Published: 15 January 2016 Academic Editor: Gregory Schneider 1 Department of Optoelectronics, Silesian University of Technology, 2 Akademicka Str., Gliwice 44-100, Poland; [email protected] (T.P.); [email protected] (M.S.) 2 Institute for Chemical Processing of Coal, 1 Zamkowa Str., Zabrze 41-803, Poland; [email protected] (R.M.); [email protected] (M.K.-M.) 3 Department of Physical Chemistry and Technology of Polymers, Silesian University of Technology, 9 Strzody Str., Gliwice 44-100, Poland; [email protected] * Correspondence: [email protected]; Tel.: +48-32-237-1263 Abstract: The paper presents the results of investigations on resistance structures based on graphite oxide (GRO) and graphene oxide (rGO). The subject matter of the investigations was thaw the sensitivity of the tested structures was affected by hydrogen, nitrogen dioxide and carbon dioxide. The experiments were performed at a temperature range from 30 ˝C to 150 ˝C in two carrier gases: nitrogen and synthetic air. The measurements were also aimed at characterization of the graphite oxide and graphene oxide. In our measurements we used (among others) techniques such as: Atomic Force Microscopy (AFM); Scanning Electron Microscopy (SEM); Raman Spectroscopy (RS); Fourier Transform Infrared Spectroscopy (FT-IR) and X-ray Photoelectron Microscopy (XPS). The data resulting from the characterizations of graphite oxide and graphene oxide have made it possible to interpret the obtained results from the point of view of physicochemical changes occurring in these structures. -

The Role of the Oxidation and Reduction Parameters on the Properties of the Reduced Graphene Oxide
coatings Article The Role of the Oxidation and Reduction Parameters on the Properties of the Reduced Graphene Oxide Marta Sieradzka * , Czesław Slusarczyk´ , Włodzimierz Binia´sand Ryszard Fryczkowski Faculty of Materials, Civil and Environmental Engineering, University of Bielsko-Biala, 43-309 Bielsko-Biala, Poland; [email protected] (C.S.);´ [email protected] (W.B.); [email protected] (R.F.) * Correspondence: [email protected] Abstract: One of the methods of obtaining reduced graphene oxide (rGO) involves the oxidation of graphite to graphene oxide, which is then exfoliated and reduced. Each of these stages has a decisive influence on the properties of the produced nanoadditive, which determines its subsequent application. The process conditions which are examined during the oxidation stage are related to: The mixing time of the reactants before oxidation, sonication of the reaction mixture, and its composition. During reduction optimization, in turn, the form of the GO sample and the method of its purification, as well as the temperature at which this process took place, are examined. At each stage, the determined structural parameters of the produced materials (GO and rGO) are related to their morphology (SEM—scanning electron microscope), oxidation state (FTIR—Fourier transform infrared spectroscopy, EDS—energy-dispersive spectrometer), structure defect (Raman spectroscopy), as well as the number of layers and crystalline structure (WAXS—wide-angle X-ray scattering). The obtained results show that the shorter mixing time of the reactants determines the formation of more oxygen functional groups. On the basis of the obtained results, the process conditions that enable the production of multilayer, well-exfoliated reduced graphene oxide, with only a slightly defected ´ Citation: Sieradzka, M.; Slusarczyk, structure, are established. -
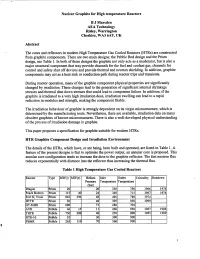
Nuclear Graphite for High Temperature Reactors
Nuclear Graphite for High temperature Reactors B J Marsden AEA Technology Risley, Warrington Cheshire, WA3 6AT, UK Abstract The cores and reflectors in modem High Temperature Gas Cooled Reactors (HTRs) are constructed from graphite components. There are two main designs; the Pebble Bed design and the Prism design, see Table 1. In both of these designs the graphite not only acts as a moderator, but is also a major structural component that may provide channels for the fuel and coolant gas, channels for control and safety shut off devices and provide thermal and neutron shielding. In addition, graphite components may act as a heat sink or conduction path during reactor trips and transients. During reactor operation, many of the graphite component physical properties are significantly changed by irradiation. These changes lead to the generation of significant internal shrinkage stresses and thermal shut down stresses that could lead to component failure. In addition, if the graphite is irradiated to a very high irradiation dose, irradiation swelling can lead to a rapid reduction in modulus and strength, making the component friable. The irradiation behaviour of graphite is strongly dependent on its virgin microstructure, which is determined by the manufacturing route. Nevertheless, there are available, irradiation data on many obsolete graphites of known microstructures. There is also a well-developed physical understanding of the process of irradiation damage in graphite. This paper proposes a specification for graphite suitable for modem HTRs. HTR Graphite Component Design and Irradiation Environment The details of the HTRs, which have, or are being, been built and operated, are listed in Table 1.