Tophat-Fusion: an Algorithm for Discovery of Novel Fusion Transcripts Daehwan Kim1* and Steven L Salzberg1,2,3
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Download Download
Supplementary Figure S1. Results of flow cytometry analysis, performed to estimate CD34 positivity, after immunomagnetic separation in two different experiments. As monoclonal antibody for labeling the sample, the fluorescein isothiocyanate (FITC)- conjugated mouse anti-human CD34 MoAb (Mylteni) was used. Briefly, cell samples were incubated in the presence of the indicated MoAbs, at the proper dilution, in PBS containing 5% FCS and 1% Fc receptor (FcR) blocking reagent (Miltenyi) for 30 min at 4 C. Cells were then washed twice, resuspended with PBS and analyzed by a Coulter Epics XL (Coulter Electronics Inc., Hialeah, FL, USA) flow cytometer. only use Non-commercial 1 Supplementary Table S1. Complete list of the datasets used in this study and their sources. GEO Total samples Geo selected GEO accession of used Platform Reference series in series samples samples GSM142565 GSM142566 GSM142567 GSM142568 GSE6146 HG-U133A 14 8 - GSM142569 GSM142571 GSM142572 GSM142574 GSM51391 GSM51392 GSE2666 HG-U133A 36 4 1 GSM51393 GSM51394 only GSM321583 GSE12803 HG-U133A 20 3 GSM321584 2 GSM321585 use Promyelocytes_1 Promyelocytes_2 Promyelocytes_3 Promyelocytes_4 HG-U133A 8 8 3 GSE64282 Promyelocytes_5 Promyelocytes_6 Promyelocytes_7 Promyelocytes_8 Non-commercial 2 Supplementary Table S2. Chromosomal regions up-regulated in CD34+ samples as identified by the LAP procedure with the two-class statistics coded in the PREDA R package and an FDR threshold of 0.5. Functional enrichment analysis has been performed using DAVID (http://david.abcc.ncifcrf.gov/) -

Early Growth Response 1 Regulates Hematopoietic Support and Proliferation in Human Primary Bone Marrow Stromal Cells
Hematopoiesis SUPPLEMENTARY APPENDIX Early growth response 1 regulates hematopoietic support and proliferation in human primary bone marrow stromal cells Hongzhe Li, 1,2 Hooi-Ching Lim, 1,2 Dimitra Zacharaki, 1,2 Xiaojie Xian, 2,3 Keane J.G. Kenswil, 4 Sandro Bräunig, 1,2 Marc H.G.P. Raaijmakers, 4 Niels-Bjarne Woods, 2,3 Jenny Hansson, 1,2 and Stefan Scheding 1,2,5 1Division of Molecular Hematology, Department of Laboratory Medicine, Lund University, Lund, Sweden; 2Lund Stem Cell Center, Depart - ment of Laboratory Medicine, Lund University, Lund, Sweden; 3Division of Molecular Medicine and Gene Therapy, Department of Labora - tory Medicine, Lund University, Lund, Sweden; 4Department of Hematology, Erasmus MC Cancer Institute, Rotterdam, the Netherlands and 5Department of Hematology, Skåne University Hospital Lund, Skåne, Sweden ©2020 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2019.216648 Received: January 14, 2019. Accepted: July 19, 2019. Pre-published: August 1, 2019. Correspondence: STEFAN SCHEDING - [email protected] Li et al.: Supplemental data 1. Supplemental Materials and Methods BM-MNC isolation Bone marrow mononuclear cells (BM-MNC) from BM aspiration samples were isolated by density gradient centrifugation (LSM 1077 Lymphocyte, PAA, Pasching, Austria) either with or without prior incubation with RosetteSep Human Mesenchymal Stem Cell Enrichment Cocktail (STEMCELL Technologies, Vancouver, Canada) for lineage depletion (CD3, CD14, CD19, CD38, CD66b, glycophorin A). BM-MNCs from fetal long bones and adult hip bones were isolated as reported previously 1 by gently crushing bones (femora, tibiae, fibulae, humeri, radii and ulna) in PBS+0.5% FCS subsequent passing of the cell suspension through a 40-µm filter. -

Identification of Novel Genes in BRCA1-Regulated Pathways
Identification of Novel Genes in BRCA1-Regulated Pathways DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Shweta Kotian, M.S. Graduate Program in Molecular, Cellular and Developmental Biology The Ohio State University 2013 Dissertation Committee: Professor Jeffrey D. Parvin, Advisor Professor Joanna L. Groden Professor Denis C. Guttridge Professor Mark R. Parthun Copyright by Shweta Kotian 2013 Abstract BRCA1 is an important breast- and ovarian-specific tumor suppressor gene. It is important in various cellular functions in the body including transcriptional regulation, cell cycle checkpoint activation, DNA damage response, and maintenance of genomic stability. Approximately 40% of hereditary breast cancers have mutations in BRCA1 or BRCA2, and it is unclear how the remaining cases are caused, presumably by mutations or in the alteration of expression of unknown genes. We hypothesize that these unknown genes or ‘missing BRCAs’ can be identified by bio-informatics methods. Performing gene co-expression analysis, we have compared the expression profiles of hundreds of genes across publicly available breast cancer microarray datasets, with those of BRCA1 and BRCA2. We propose that the genes, whose expression is highly correlated with BRCA1 and BRCA2, might function together in the same pathways. They could also potentially interact with each other. The main aim of this study is to identify these genes, and then biologically validate them in functional BRCA1-regulated pathways including homologous recombination and centrosome duplication. We also hope to find any mechanistic ii associations between these genes and BRCA1. Eventually, we hope to evaluate if these genes contribute to the process of carcinogenesis. -

Datasheet Blank Template
SANTA CRUZ BIOTECHNOLOGY, INC. WIPI-3 siRNA (h): sc-72214 BACKGROUND STORAGE AND RESUSPENSION WD-repeats are motifs that are found in a variety of proteins and are charac- Store lyophilized siRNA duplex at -20° C with desiccant. Stable for at least terized by a conserved core of 40-60 amino acids that commonly form a terti- one year from the date of shipment. Once resuspended, store at -20° C, ary propeller structure. While proteins that contain WD-repeats participate in avoid contact with RNAses and repeated freeze thaw cycles. a wide range of cellular functions, they are generally involved in regulatory Resuspend lyophilized siRNA duplex in 330 µl of the RNAse-free water mechanisms concerning chromatin assembly, cell cycle control, signal trans- provided. Resuspension of the siRNA duplex in 330 µl of RNAse-free water duction, RNA processing, apoptosis and vesicular trafficking. WIPI-3 (WD makes a 10 µM solution in a 10 µM Tris-HCl, pH 8.0, 20 mM NaCl, 1 mM repeat domain phosphoinositide-interacting protein 3), also known as WD EDTA buffered solution. repeat-containing protein 45-like or WIPI49-like protein, is a 344 amino acid protein that is ubiquitously expressed with highest levels found in pancreas, APPLICATIONS heart and skeletal muscle. Upregulated in uterine and ovarian cancer, WIPI-3 contains two WD domains, seven WD40 domains, and is encoded by a gene ARMER siRNA (m) is recommended for the inhibition of ARMER expression that maps to human chromosome 17q25.3. Chromosome 17 comprises over in mouse cells. 2.5% of the human genome, encodes over 1,200 genes and is associated with two key tumor suppressor genes, namely, p53 and BRCA1. -

Coexpression Networks Based on Natural Variation in Human Gene Expression at Baseline and Under Stress
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations Fall 2010 Coexpression Networks Based on Natural Variation in Human Gene Expression at Baseline and Under Stress Renuka Nayak University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Computational Biology Commons, and the Genomics Commons Recommended Citation Nayak, Renuka, "Coexpression Networks Based on Natural Variation in Human Gene Expression at Baseline and Under Stress" (2010). Publicly Accessible Penn Dissertations. 1559. https://repository.upenn.edu/edissertations/1559 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/1559 For more information, please contact [email protected]. Coexpression Networks Based on Natural Variation in Human Gene Expression at Baseline and Under Stress Abstract Genes interact in networks to orchestrate cellular processes. Here, we used coexpression networks based on natural variation in gene expression to study the functions and interactions of human genes. We asked how these networks change in response to stress. First, we studied human coexpression networks at baseline. We constructed networks by identifying correlations in expression levels of 8.9 million gene pairs in immortalized B cells from 295 individuals comprising three independent samples. The resulting networks allowed us to infer interactions between biological processes. We used the network to predict the functions of poorly-characterized human genes, and provided some experimental support. Examining genes implicated in disease, we found that IFIH1, a diabetes susceptibility gene, interacts with YES1, which affects glucose transport. Genes predisposing to the same diseases are clustered non-randomly in the network, suggesting that the network may be used to identify candidate genes that influence disease susceptibility. -
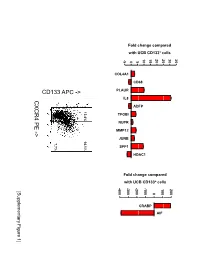
Supplementary Data
Fold change compared with UCB CD133+ cells 10 15 20 25 30 35 -5 0 5 COL4A1 CD68 PLAUR IL8 CXCR4 PE ADFP 13.4% 14.1% TFGBI PE NUPR - MMP12 > JUNB 7.7% SPP1 HDAC1 Fold change compared with UCB CD133+ cells -400 -300 -200 -100 100 200 [Supplementary Figure1] 0 CRABP AIF Rutella S et al. Supplementary Materials and Methods Real-time PCR Real-time quantitative PCR (qPCR) was performed on mRNA samples isolated from either endometrial cancer-derived CD133+ cells or umbilical cord blood CD133+ cells with the RNeasy plus Mini Kit (QIAGEN, Hilden, Germany), as already detailed. Quantitative PCR for a selected group of genes was performed on the iCycler iQ system (Bio-Rad, Hercules, CA). Primer sets were designed using the Beacon Design Software (Version 3) and the sequences available in the GeneBankTM data base. The specific oligonucleotide primer sequences are detailed in Supplementary Table 8. Complementary DNA (cDNA) was prepared starting from 1μg of total RNA using the iScrypt cDNA Synthesis Kit, which contains RNase H + MMLV reverse transcriptase random primers and 5x reaction mixture (Bio-Rad), according to the manufacturer’s instructions. Reactions were conducted in the PTC-0200 DNA Engine (MJ RESEARCH). Amplification was carried out in a total volume of 25μl containing 0.3μM of each specific primer, 12,5μl 2X SYBR Green Master Mix (100mM KCl, 40mM Tris-HCl, pH 8.4, 0.4mM of each dNTP, iTaq DNA polymerase, 6mM MgCl2, SYBR Green I, 20nM fluorescein, stabilizers; Bio-Rad) and 2μl of diluted cDNA. The PCR reactions were cycled starting with a 3-minute template denaturation step at 95°C followed by 40 cycles of 15 seconds at 95°C and 1 minute at 60°C. -

Table S8: Summary of CN Loss Genes Identified by Fisher Test on SNP-FASST2 Segmentation
Table S8: Summary of CN Loss Genes identified by Fisher Test on SNP-FASST2 Segmentation Gene Symbol Chromosome Start End Length SLC6A19 chr5 1201709 1225230 23522 SLC6A18 chr5 1225469 1246304 20836 TERT chr5 1253286 1295162 41877 SURF6 chr9 136197551 136203047 5497 MED22 chr9 136207754 136214972 7219 SNORD24 chr9 136216250 136216325 76 RPL7A chr9 136215068 136218280 3213 SNORD36B chr9 136216948 136217019 72 SNORD36A chr9 136217310 136217382 73 SNORD36C chr9 136217700 136217767 68 SURF1 chr9 136218665 136223361 4697 SURF2 chr9 136223420 136228040 4621 SURF4 chr9 136228339 136242970 14632 C9orf96 chr9 136243283 136271220 27938 REXO4 chr9 136271185 136283164 11980 ADAMTS13 chr9 136279458 136324508 45051 CACFD1 chr9 136325086 136335909 10824 SLC2A6 chr9 136336215 136344276 8062 TMEM8C chr9 136379707 136390068 10362 ADAMTSL2 chr9 136397285 136440641 43357 FAM163B chr9 136443536 136445368 1833 DBH chr9 136501484 136524466 22983 SARDH chr9 136528683 136605077 76395 VAV2 chr9 136627015 136857446 230432 LINC00094 chr9 136890560 136896719 6160 BRD3 chr9 136895445 136933141 37697 WDR5 chr9 137001209 137025094 23886 RNU6ATAC chr9 137029561 137029686 126 OLFM1 chr9 137967088 138013030 45943 LOC401557 chr9 138073454 138079152 5699 DDX11L10 chr16 61554 64090 2537 POLR3K chr16 96978 103632 6655 SNRNP25 chr16 103828 107669 3842 RHBDF1 chr16 108057 122629 14573 MPG chr16 127017 135850 8834 NPRL3 chr16 135803 188697 52895 HBZ chr16 202853 204504 1652 HBM chr16 215972 216767 796 HBA2 chr16 222845 223709 865 HBA1 chr16 226678 227520 843 HBQ1 chr16 230332 231178 -

Supplementary Table 2. 503-‐Gene List Differentially Expressed
Supplementary Table 2. 503-gene list differentially expressed between pPCL and MM. Fold Gene Symbol Gene Name Cytoband Score(d) Change FLNA filamin A, alpha (actin binding protein 280) Xq28 8.8939 4.3943 TAGLN2 transgelin 2 1q21-q25 6.9247 3.1111 USP36 ubiquitin specific peptidase 36 17q25.3 6.7445 1.5881 ADAM8 ADAM metallopeptidase domain 8 10q26.3 6.7222 2.0512 CORO7 coronin 7 16p13.3 6.4958 2.4215 FAS Fas (TNF receptor superfamily, member 6) 10q24.1 6.3575 4.2037 FMNL1 formin-like 1 17q21 6.2702 1.8204 TNIP2 TNFAIP3 interacting protein 2 4p16.3 6.2603 2.0729 CD99 CD99 molecule Xp22.32;Yp11.3 6.2291 6.7692 ANXA11 annexin A11 10q23 6.0995 2.3510 CYB5R3 cytochrome b5 reductase 3 22q13.2-q13.31;22q13.31-qter 6.0525 1.6998 ARHGDIA Rho GDP dissociation inhibitor (GDI) alpha 17q25.3 5.9989 1.6278 KIF21B kinesin family member 21B 1pter-q31.3 5.8725 2.3523 CCR7 chemokine (C-C motif) receptor 7 17q12-q21.2 5.8384 2.7242 SMPD4 sphingomyelin phosphodiesterase 4, neutral membrane (neutral sphingomyelinase-3) 2q21.1 5.8162 1.5613 NDUFA6 NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 6, 14kDa 22q13.2 5.8079 1.7231 EMP3 epithelial membrane protein 3 19q13.3 5.8040 2.5566 KHDRBS1 KH domain containing, RNA binding, signal transduction associated 1 1p32 5.7410 1.4743 TRAF5 TNF receptor-associated factor 5 1q32 5.7267 2.5907 WDR1 WD repeat domain 1 4p16.1 5.6831 1.8278 LGALS3 lectin, galactoside-binding, soluble, 3 14q22.3 5.6669 2.1458 HABP4 hyaluronan binding protein 4 9q22.3-q31 5.6466 2.2176 C6orf1 chromosome 6 open reading frame 1 6p21.31 5.6343 -

Identification and Mechanistic Investigation of Recurrent Functional Genomic and Transcriptional Alterations in Advanced Prostat
Identification and Mechanistic Investigation of Recurrent Functional Genomic and Transcriptional Alterations in Advanced Prostate Cancer Thomas A. White A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Washington 2013 Reading Committee: Peter S. Nelson, Chair Janet L. Stanford Raymond J. Monnat Jay A. Shendure Program Authorized to Offer Degree: Molecular and Cellular Biology ©Copyright 2013 Thomas A. White University of Washington Abstract Identification and Mechanistic Investigation of Recurrent Functional Genomic and Transcriptional Alterations in Advanced Prostate Cancer Thomas A. White Chair of the Supervisory Committee: Peter S. Nelson, MD Novel functionally altered transcripts may be recurrent in prostate cancer (PCa) and may underlie lethal and advanced disease and the neuroendocrine small cell carcinoma (SCC) phenotype. We conducted an RNASeq survey of the LuCaP series of 24 PCa xenograft tumors from 19 men, and validated observations on metastatic tumors and PCa cell lines. Key findings include discovery and validation of 40 novel fusion transcripts including one recurrent chimera, many SCC-specific and castration resistance (CR) -specific novel splice isoforms, new observations on SCC-specific and TMPRSS2-ERG specific differential expression, the allele-specific expression of certain recurrent non- synonymous somatic single nucleotide variants (nsSNVs) previously discovered via exome sequencing of the same tumors, rgw ubiquitous A-to-I RNA editing of base excision repair (BER) gene NEIL1 as well as CDK13, a kinase involved in RNA splicing, and the SCC expression of a previously unannotated long noncoding RNA (lncRNA) at Chr6p22.2. Mechanistic investigation of the novel lncRNA indicates expression is regulated by derepression by master neuroendocrine regulator RE1-silencing transcription factor (REST), and may regulate some genes of axonogenesis and angiogenesis. -

1 Imipramine Treatment and Resiliency Exhibit Similar
Imipramine Treatment and Resiliency Exhibit Similar Chromatin Regulation in the Mouse Nucleus Accumbens in Depression Models Wilkinson et al. Supplemental Material 1. Supplemental Methods 2. Supplemental References for Tables 3. Supplemental Tables S1 – S24 SUPPLEMENTAL TABLE S1: Genes Demonstrating Increased Repressive DimethylK9/K27-H3 Methylation in the Social Defeat Model (p<0.001) SUPPLEMENTAL TABLE S2: Genes Demonstrating Decreased Repressive DimethylK9/K27-H3 Methylation in the Social Defeat Model (p<0.001) SUPPLEMENTAL TABLE S3: Genes Demonstrating Increased Repressive DimethylK9/K27-H3 Methylation in the Social Isolation Model (p<0.001) SUPPLEMENTAL TABLE S4: Genes Demonstrating Decreased Repressive DimethylK9/K27-H3 Methylation in the Social Isolation Model (p<0.001) SUPPLEMENTAL TABLE S5: Genes Demonstrating Common Altered Repressive DimethylK9/K27-H3 Methylation in the Social Defeat and Social Isolation Models (p<0.001) SUPPLEMENTAL TABLE S6: Genes Demonstrating Increased Repressive DimethylK9/K27-H3 Methylation in the Social Defeat and Social Isolation Models (p<0.001) SUPPLEMENTAL TABLE S7: Genes Demonstrating Decreased Repressive DimethylK9/K27-H3 Methylation in the Social Defeat and Social Isolation Models (p<0.001) SUPPLEMENTAL TABLE S8: Genes Demonstrating Increased Phospho-CREB Binding in the Social Defeat Model (p<0.001) SUPPLEMENTAL TABLE S9: Genes Demonstrating Decreased Phospho-CREB Binding in the Social Defeat Model (p<0.001) SUPPLEMENTAL TABLE S10: Genes Demonstrating Increased Phospho-CREB Binding in the Social -

Supplementary Data Materials And
Supplementary Data Materials and methods Cell culture HepG2 human hepatoblastoma cells were obtained from ATCC (HB-8065, Rockville, MD, USA). Cells were grown in a humidified incubator (20% O2, 5% CO2 at 37°C) (Sanyo MCO-18M O2/CO2 incubator, Osaka, Japan) in Williams Medium E (WEM, InVitrogen) supplemented with 10% fetal calf serum, 2 mM L- glutamine, 20 mU/ml insulin, 50 nM dexamethasone, 100 U/ml penicillin, 100 µg/ml streptomycin, 2.5 µg fungizone, 50 µg/ml gentamycin and 100 µg/ml vancomycin (=WEM-C). For the microarray analysis two experiments were executed in parallel. Cells 6 were seeded at 3x10 in 75 cm² tissue culture flasks (n=4) at 20% O2 and were grown until 70% confluence. After reaching near-confluence, 2 flasks were placed in a humidified incubator with hypoxic conditions (2% O2, 5% CO2 at 37°C), while the other flasks (n=2) remained in normoxic conditions (20% O2). Cells were cultured for 72hrs in these different oxygen conditions and after three days cells were harvested after trypsin treatment, mixed with Trizol (InVitrogen, Merelbeke, Belgium) and stored in -80°C for further analysis. Sample Collection and Microarray Target Synthesis and Processing Samples in Trizol were homogenized in a Dounce homogenizer for RNA extraction. Thereafter, RNA was isolated with the RNeasy Kit (Qiagen, Chatsworth, CA) according to the manufacturer’s instructions. The quality of all RNA samples was monitored with a NanoDrop spectrophotometer (NanoDrop Technologies, Centreville, DE) and by means of the Agilent 2100 BioAnalyzer (Agilent, Palo Alto, CA). Only RNA showing no signs of degradation or impurities (260/280 and 260/230 nm ratios, >1.8) was considered suitable for microarray analysis and used for labeling.