The Role of C-Myc in Regulation of Translation Initiation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Host Cell Factors Necessary for Influenza a Infection: Meta-Analysis of Genome Wide Studies
Host Cell Factors Necessary for Influenza A Infection: Meta-Analysis of Genome Wide Studies Juliana S. Capitanio and Richard W. Wozniak Department of Cell Biology, Faculty of Medicine and Dentistry, University of Alberta Abstract: The Influenza A virus belongs to the Orthomyxoviridae family. Influenza virus infection occurs yearly in all countries of the world. It usually kills between 250,000 and 500,000 people and causes severe illness in millions more. Over the last century alone we have seen 3 global influenza pandemics. The great human and financial cost of this disease has made it the second most studied virus today, behind HIV. Recently, several genome-wide RNA interference studies have focused on identifying host molecules that participate in Influen- za infection. We used nine of these studies for this meta-analysis. Even though the overlap among genes identified in multiple screens was small, network analysis indicates that similar protein complexes and biological functions of the host were present. As a result, several host gene complexes important for the Influenza virus life cycle were identified. The biological function and the relevance of each identified protein complex in the Influenza virus life cycle is further detailed in this paper. Background and PA bound to the viral genome via nucleoprotein (NP). The viral core is enveloped by a lipid membrane derived from Influenza virus the host cell. The viral protein M1 underlies the membrane and anchors NEP/NS2. Hemagglutinin (HA), neuraminidase Viruses are the simplest life form on earth. They parasite host (NA), and M2 proteins are inserted into the envelope, facing organisms and subvert the host cellular machinery for differ- the viral exterior. -

Overview of Research on Fusion Genes in Prostate Cancer
2011 Review Article Overview of research on fusion genes in prostate cancer Chunjiao Song1,2, Huan Chen3 1Medical Research Center, Shaoxing People’s Hospital, Shaoxing University School of Medicine, Shaoxing 312000, China; 2Shaoxing Hospital, Zhejiang University School of Medicine, Shaoxing 312000, China; 3Key Laboratory of Microorganism Technology and Bioinformatics Research of Zhejiang Province, Zhejiang Institute of Microbiology, Hangzhou 310000, China Contributions: (I) Conception and design: C Song; (II) Administrative support: Shaoxing Municipal Health and Family Planning Science and Technology Innovation Project (2017CX004) and Shaoxing Public Welfare Applied Research Project (2018C30058); (III) Provision of study materials or patients: None; (IV) Collection and assembly of data: C Song; (V) Data analysis and interpretation: H Chen; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. Correspondence to: Chunjiao Song. No. 568 Zhongxing Bei Road, Shaoxing 312000, China. Email: [email protected]. Abstract: Fusion genes are known to drive and promote carcinogenesis and cancer progression. In recent years, the rapid development of biotechnologies has led to the discovery of a large number of fusion genes in prostate cancer specimens. To further investigate them, we summarized the fusion genes. We searched related articles in PubMed, CNKI (Chinese National Knowledge Infrastructure) and other databases, and the data of 92 literatures were summarized after preliminary screening. In this review, we summarized approximated 400 fusion genes since the first specific fusion TMPRSS2-ERG was discovered in prostate cancer in 2005. Some of these are prostate cancer specific, some are high-frequency in the prostate cancer of a certain ethnic group. This is a summary of scientific research in related fields and suggests that some fusion genes may become biomarkers or the targets for individualized therapies. -

Supplemental Material For
SUPPLEMENTAL MATERIAL FOR Coexpression network based on natural variation in human gene expression reveals gene interactions and functions Renuka Nayak, Michael Kearns, Richard S. Spielman, Vivian G. Cheung Supplementary Figure 1 Supplementary Table 1. Gene pairs whose correlations in gene expression levels differ significantly (Pc<0.05) among the 3 datasets. Supplementary Table 2. Gene pairs that are correlated in gene expression levels with |R|>0.5 and are found within 500 kb of each other. Supplementary Table 3. Predicted functions of poorly characterized genes based on the functions of neighboring genes. Supplementary Figure 1. Genes identified in genome-wide association studies (grey) and their neighbors in the network. Red and green connections refer to positive and negative correlations, respectively. MICB has been implicated in AIDS progression (PMID: 19115949) TNF has been implicated in AIDS progression (PMID: 19115949) LTB has been implicated in AIDS progression (PMID: 19115949) ZNF224 has been implicated in Alzheimer's disease (PMID: 19118814) NDUFAB1 has been implicated in bipolar disorder (PMID: 17554300) SFRS10 has been implicated in body mass index (PMID: 19079260) and weight (PMID: 19079260) CTNNBL1 has been implicated in bone mineral density (PMID: 17903296) TGFBR3 has been implicated in bone mineral density (PMID: 19249006) IGF2R has been implicated in brain lesion load (PMID: 19010793) LSP1 has been implicated in breast cancer (PMID: 17529967) FBN1 has been implicated in breast cancer (PMID: 17903305) GLG1 has been implicated in breast cancer (PMID: 18463975) SCHIP1 has been implicated in Celiac disease (PMID: 18311140) RGS1 has been implicated in Celiac disease (PMID: 18311140) FADS2 has been implicated in Cholesterol (total) (PMID: 19060911), HDL cholesterol (PMID: 19060911, 19060906), LDL cholesterol (PMID: 19060911, 19060910), and triglycerides (PMID: 19060906). -

The Role of Eif5a Hypusination in Mediating Oncogenic Mtor Signaling
The role of eIF5A hypusination in mediating oncogenic mTOR signaling Yutian Cai Faculty of Medicine Department of Biochemistry McGill University, Montreal, Quebec, Canada December 2016 A thesis submitted to McGill University in partial fulfillment of the requirements of the degree of Master of Science ©Yutian Cai 2016 TABLE OF CONTENTS TABLE OF CONTENTS ........................................................................................ 2 TABLE OF FIGURES ............................................................................................ 4 PREFACE.............................................................................................................. 5 ACKNOWLEDGEMENTS ...................................................................................... 6 ABSTRACT ........................................................................................................... 7 RÉSUMÉ ............................................................................................................... 9 LIST OF ABBREVIATIONS ................................................................................. 11 CHAPTER 1: INTRODUCTION ........................................................................... 14 1.1 mRNA TRANSLATION ......................................................................... 14 1.1.1 TRANSLATION INITIATION....................................................................... 15 1.1.2 TRANSLATIONAL EFFICIENCY ............................................................... 20 1.1.3 TRANSLATION AND CANCER -

Autocrine IFN Signaling Inducing Profibrotic Fibroblast Responses By
Downloaded from http://www.jimmunol.org/ by guest on September 23, 2021 Inducing is online at: average * The Journal of Immunology , 11 of which you can access for free at: 2013; 191:2956-2966; Prepublished online 16 from submission to initial decision 4 weeks from acceptance to publication August 2013; doi: 10.4049/jimmunol.1300376 http://www.jimmunol.org/content/191/6/2956 A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Autocrine IFN Signaling Feng Fang, Kohtaro Ooka, Xiaoyong Sun, Ruchi Shah, Swati Bhattacharyya, Jun Wei and John Varga J Immunol cites 49 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130037 6.DC1 This article http://www.jimmunol.org/content/191/6/2956.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 23, 2021. The Journal of Immunology A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Inducing Autocrine IFN Signaling Feng Fang,* Kohtaro Ooka,* Xiaoyong Sun,† Ruchi Shah,* Swati Bhattacharyya,* Jun Wei,* and John Varga* Activation of TLR3 by exogenous microbial ligands or endogenous injury-associated ligands leads to production of type I IFN. -

Upstream Open Reading Frames Differentially Regulate Gene
UPSTREAM OPEN READING FRAMES DIFFERENTIALLY REGULATE GENE- SPECIFIC TRANSLATION IN THE INTEGRATED STRESS RESPONSE Sara Kathryn Young Submitted to the faculty of the University Graduate School in partial fulfillment of the requirements for the degree Doctor of Philosophy in the Department of Biochemistry and Molecular Biology Indiana University July 2016 Accepted by the Graduate Faculty, Indiana University, in partial fulfillment of the requirements for the degree of Doctor of Philosophy. ____________________________________ Ronald C. Wek, Ph.D., Chair ____________________________________ Murray Korc, M.D. Doctoral Committee ____________________________________ Amber L. Mosley, Ph.D. May 13, 2016 ____________________________________ John J. Turchi, Ph.D. ii © 2016 Sara Kathryn Young iii ACKNOWLEDGEMENTS I am foremost indebted to Dr. Ronald C. Wek for being an extraordinary mentor who has worked to mold me into a capable research scientist. I appreciate the time and dedication of my research committee: Dr. Murray Korc, Dr. Amber L. Mosley, and Dr. John J. Turchi. Past and current members of the Wek lab were paramount to the work presented in this thesis, particularly Dr. Thomas D. Baird with his invaluable contribution to the EPRS project and overall support. I would also like to the thank the faculty and staff of the Department of Biochemistry for their generous support and guidance. This work was supported by National Institutes of Health Grant GM049164 to R.C.W. iv Sara Kathryn Young UPSTREAM OPEN READING FRAMES DIFFERENTIALLY REGULATE GENE- SPECIFIC TRANSLATION IN THE INTEGRATED STRESS RESPONSE Gene expression is a highly coordinated process that relies upon appropriate regulation of translation for protein homeostasis. Regulation of protein synthesis largely occurs at the initiation step in which the translational start site is selected by ribosomes and associated initiating factors. -
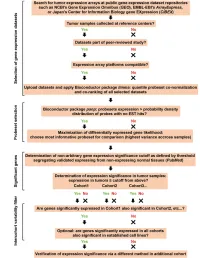
Supplementary Data
SUPPLEMENTAL INFORMATION A study restricted to chemokine receptors as well as a genome-wide transcript analysis uncovered CXCR4 as preferentially expressed in Ewing's sarcoma (Ewing's sarcoma) cells of metastatic origin (Figure 4). Transcriptome analyses showed that in addition to CXCR4, genes known to support cell motility and invasion topped the list of genes preferentially expressed in metastasis-derived cells (Figure 4D). These included kynurenine 3-monooxygenase (KMO), galectin-1 (LGALS1), gastrin-releasing peptide (GRP), procollagen C-endopeptidase enhancer (PCOLCE), and ephrin receptor B (EPHB3). KMO, a key enzyme of tryptophan catabolism, has not been linked to metastasis. Tryptophan and its catabolites, however, are involved in immune evasion by tumors, a process that can assist in tumor progression and metastasis (1). LGALS1, GRP, PCOLCE and EPHB3 have been linked to tumor progression and metastasis of several cancers (2-4). Top genes preferentially expressed in L-EDCL included genes that suppress cell motility and/or potentiate cell adhesion such as plakophilin 1 (PKP1), neuropeptide Y (NPY), or the metastasis suppressor TXNIP (5-7) (Figure 4D). Overall, L-EDCL were enriched in gene sets geared at optimizing nutrient transport and usage (Figure 4D; Supplementary Table 3), a state that may support the early stages of tumor growth. Once tumor growth outpaces nutrient and oxygen supplies, gene expression programs are usually switched to hypoxic response and neoangiogenesis, which ultimately lead to tumor egress and metastasis. Accordingly, gene sets involved in extracellular matrix remodeling, MAPK signaling, and response to hypoxia were up-regulated in M-EDCL (Figure 4D; Supplementary Table 4), consistent with their association to metastasis in other cancers (8, 9). -

New Pancreatic Cancer Biomarkers Eif1, Eif2d, Eif3c and Eif6 Play A
ANTICANCER RESEARCH 40 : 3109-3118 (2020) doi:10.21873/anticanres.14292 New Pancreatic Cancer Biomarkers eIF1, eIF2D, eIF3C and eIF6 Play a Major Role in Translational Control in Ductal Adenocarcinoma NICOLE GOLOB-SCHWARZL 1,2 , PHILIP PUCHAS 1, MARGIT GOGG-KAMERER 1, WILKO WEICHERT 3, BENJAMIN GÖPPERT 4 and JOHANNES HAYBAECK 1,5 1Diagnostic and Research Institute of Pathology, Medical University of Graz, Graz, Austria; 2Institute of Dermatology and Venerology, Medical University of Graz, Graz, Austria; 3Institute of Pathology, Technical University Munich, Munich, Germany; 4Department of General Pathology and Anatomy of the Pathology Institute, University Hospital Heidelberg, Heidelberg, Germany; 5Department of Pathology, Neuropathology and Molecular Pathology, Medical University of Innsbruck, Innsbruck, Austria Abstract. Background/Aim: Pancreatic cancer is one of the Consequently, they might be useful as potential new deadliest forms of cancer and ranks among the leading causes biomarkers and therapeutic targets in PDAC. of cancer-related death worldwide. The most common histological type is ductal adenocarcinoma (PDAC), Pancreatic cancer, including ductal adenocarcinoma (PDAC), accounting for approximately 95% of cases. Deregulation of its most common manifestation, is still one of the deadliest protein synthesis has been found to be closely related to cancer. forms of cancer. It usually affects the elderly, with a 5-year The rate-limiting step of translation is initiation, which is survival rate of 5% that has not improved over the past 20 regulated by a broad range of eukaryotic translation initiation years (1). PDAC, originates from the exocrine portion of the factors (eIFs). Patients and Methods: Human PDAC samples gland and accounts for approximately 80% of cases (1). -

SUPPLEMENTARY MATERIALS and METHODS PBMC Transcriptomics
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) Gut SUPPLEMENTARY MATERIALS AND METHODS PBMC transcriptomics identifies immune-metabolism disorder during the development of HBV-ACLF Contents l Supplementary methods l Supplementary Figure 1 l Supplementary Figure 2 l Supplementary Figure 3 l Supplementary Figure 4 l Supplementary Figure 5 l Supplementary Table 1 l Supplementary Table 2 l Supplementary Table 3 l Supplementary Table 4 l Supplementary Tables 5-14 l Supplementary Table 15 l Supplementary Table 16 l Supplementary Table 17 Li J, et al. Gut 2021;0:1–13. doi: 10.1136/gutjnl-2020-323395 BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) Gut SUPPLEMENTARY METHODS Test for HBV DNA The levels of HBV DNA were detected using real-time PCR with a COBAS® AmpliPrep/COBAS® TaqMan 48 System (Roche, Basel, Switzerland) and HBV Test v2.0. Criteria for diagnosing cirrhosis Pathology The gold standard for the diagnosis of cirrhosis is a liver biopsy obtained through a percutaneous or transjugular approach.1 Ultrasonography was performed 2-4 hours before biopsy. Liver biopsy specimens were obtained by experienced physicians. Percutaneous transthoracic puncture of the liver was performed according to the standard criteria. After biopsy, patients were monitored in the hospital with periodic analyses of haematocrit and other vital signs for 24 hours. Cirrhosis was diagnosed according to the globally agreed upon criteria.2 Cirrhosis is defined based on its pathological features under a microscope: (a) the presence of parenchymal nodules, (b) differences in liver cell size and appearance, (c) fragmentation of the biopsy specimen, (d) fibrous septa, and (d) an altered architecture and vascular relationships. -

Original Article Overexpression of Eif3e Is Correlated with Colon Tumor Development and Poor Prognosis
Int J Clin Exp Pathol 2014;7(10):6462-6474 www.ijcep.com /ISSN:1936-2625/IJCEP0002023 Original Article Overexpression of eIF3e is correlated with colon tumor development and poor prognosis Zhi Li1*, Shengtao Lin2*, Tao Jiang3, Jingtao Wang2, Huijun Lu4, Huamei Tang4, Mujian Teng5, Junwei Fan2 1Department of Anorectal Surgery, Qianfoshan Hospital Affiliated to Shandong University, 16766 Jingshi Road, Jinan 250014, Shandong, China; 2Department of General Surgery, Shanghai Jiaotong University Affiliated First People’s Hospital, 85 Wujin Road, Shanghai 200080, China; 3Department of Anal-Colorectal Surgery, General Hospital of Ningxia Medical University, 804 South Shengli Road, Yinchuan 750004, China; 4Department of Pathology, Shanghai Jiaotong University Affiliated First People’s Hospital, 85 Wujin Road, Shanghai 200080, China; 5Department of General Surgery, Qianfoshan Hospital Affiliated to Shandong University, 16766 Jingshi Road, Jinan 250014, Shandong, China. *Equal contributors. Received August 21, 2014; Accepted September 15, 2014; Epub September 15, 2014; Published October 1, 2014 Abstract: EIF3e is a component of the eukaryotic translation initiation factor 3 (eIF-3) complexes, which is an es- sential factor for initiation of protein synthesis in mammalian cells. Translational control plays key roles in the complex mechanism of cancer development and progression. However, the clinical significance of eIF3e in colon cancer remains to be elucidated. We analyzed the eIF3e expression in a tissue microarray (TMA), which contained 173 colon cancer tissues paired with adjacent normal mucosa and lymph node metastasis. The expression of eIF3e was significantly elevated in colon cancer tissues in comparison with those in adjacent normal mucosa (P < 0.001) and lymph node metastasis (P < 0.001). -

Novel and Highly Recurrent Chromosomal Alterations in Se´Zary Syndrome
Research Article Novel and Highly Recurrent Chromosomal Alterations in Se´zary Syndrome Maarten H. Vermeer,1 Remco van Doorn,1 Remco Dijkman,1 Xin Mao,3 Sean Whittaker,3 Pieter C. van Voorst Vader,4 Marie-Jeanne P. Gerritsen,5 Marie-Louise Geerts,6 Sylke Gellrich,7 Ola So¨derberg,8 Karl-Johan Leuchowius,8 Ulf Landegren,8 Jacoba J. Out-Luiting,1 Jeroen Knijnenburg,2 Marije IJszenga,2 Karoly Szuhai,2 Rein Willemze,1 and Cornelis P. Tensen1 Departments of 1Dermatology and 2Molecular Cell Biology, Leiden University Medical Center, Leiden, the Netherlands; 3Department of Dermatology, St Thomas’ Hospital, King’s College, London, United Kingdom; 4Department of Dermatology, University Medical Center Groningen, Groningen, the Netherlands; 5Department of Dermatology, Radboud University Nijmegen Medical Center, Nijmegen, the Netherlands; 6Department of Dermatology, Gent University Hospital, Gent, Belgium; 7Department of Dermatology, Charite, Berlin, Germany; and 8Department of Genetics and Pathology, Rudbeck Laboratory, University of Uppsala, Uppsala, Sweden Abstract Introduction This study was designed to identify highly recurrent genetic Se´zary syndrome (Sz) is an aggressive type of cutaneous T-cell alterations typical of Se´zary syndrome (Sz), an aggressive lymphoma/leukemia of skin-homing, CD4+ memory T cells and is cutaneous T-cell lymphoma/leukemia, possibly revealing characterized by erythroderma, generalized lymphadenopathy, and pathogenetic mechanisms and novel therapeutic targets. the presence of neoplastic T cells (Se´zary cells) in the skin, lymph High-resolution array-based comparative genomic hybridiza- nodes, and peripheral blood (1). Sz has a poor prognosis, with a tion was done on malignant T cells from 20 patients. disease-specific 5-year survival of f24% (1).