Microdistribution and Retention of Chloropicrin in Wood
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Field Guide to Common Macrofungi in Eastern Forests and Their Ecosystem Functions
United States Department of Field Guide to Agriculture Common Macrofungi Forest Service in Eastern Forests Northern Research Station and Their Ecosystem General Technical Report NRS-79 Functions Michael E. Ostry Neil A. Anderson Joseph G. O’Brien Cover Photos Front: Morel, Morchella esculenta. Photo by Neil A. Anderson, University of Minnesota. Back: Bear’s Head Tooth, Hericium coralloides. Photo by Michael E. Ostry, U.S. Forest Service. The Authors MICHAEL E. OSTRY, research plant pathologist, U.S. Forest Service, Northern Research Station, St. Paul, MN NEIL A. ANDERSON, professor emeritus, University of Minnesota, Department of Plant Pathology, St. Paul, MN JOSEPH G. O’BRIEN, plant pathologist, U.S. Forest Service, Forest Health Protection, St. Paul, MN Manuscript received for publication 23 April 2010 Published by: For additional copies: U.S. FOREST SERVICE U.S. Forest Service 11 CAMPUS BLVD SUITE 200 Publications Distribution NEWTOWN SQUARE PA 19073 359 Main Road Delaware, OH 43015-8640 April 2011 Fax: (740)368-0152 Visit our homepage at: http://www.nrs.fs.fed.us/ CONTENTS Introduction: About this Guide 1 Mushroom Basics 2 Aspen-Birch Ecosystem Mycorrhizal On the ground associated with tree roots Fly Agaric Amanita muscaria 8 Destroying Angel Amanita virosa, A. verna, A. bisporigera 9 The Omnipresent Laccaria Laccaria bicolor 10 Aspen Bolete Leccinum aurantiacum, L. insigne 11 Birch Bolete Leccinum scabrum 12 Saprophytic Litter and Wood Decay On wood Oyster Mushroom Pleurotus populinus (P. ostreatus) 13 Artist’s Conk Ganoderma applanatum -

A New Record of Ganoderma Tropicum (Basidiomycota, Polyporales) for Thailand and First Assessment of Optimum Conditions for Mycelia Production
A peer-reviewed open-access journal MycoKeys 51:A new65–83 record (2019) of Ganoderma tropicum (Basidiomycota, Polyporales) for Thailand... 65 doi: 10.3897/mycokeys.51.33513 RESEARCH ARTICLE MycoKeys http://mycokeys.pensoft.net Launched to accelerate biodiversity research A new record of Ganoderma tropicum (Basidiomycota, Polyporales) for Thailand and first assessment of optimum conditions for mycelia production Thatsanee Luangharn1,2,3,4, Samantha C. Karunarathna1,3,4, Peter E. Mortimer1,4, Kevin D. Hyde3,5, Naritsada Thongklang5, Jianchu Xu1,3,4 1 Key Laboratory for Plant Diversity and Biogeography of East Asia, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, China 2 University of Chinese Academy of Sciences, Bei- jing 100049, China 3 East and Central Asia Regional Office, World Agroforestry Centre (ICRAF), Kunming 650201, Yunnan, China 4 Centre for Mountain Ecosystem Studies (CMES), Kunming Institute of Botany, Kunming 650201, Yunnan, China 5 Center of Excellence in Fungal Research, Mae Fah Luang University, Chiang Rai 57100, Thailand Corresponding author: Jianchu Xu ([email protected]); Peter E. Mortimer ([email protected]) Academic editor: María P. Martín | Received 30 January 2019 | Accepted 12 March 2019 | Published 7 May 2019 Citation: Luangharn T, Karunarathna SC, Mortimer PE, Hyde KD, Thongklang N, Xu J (2019) A new record of Ganoderma tropicum (Basidiomycota, Polyporales) for Thailand and first assessment of optimum conditions for mycelia production. MycoKeys 51: 65–83. https://doi.org/10.3897/mycokeys.51.33513 Abstract In this study a new record of Ganoderma tropicum is described as from Chiang Rai Province, Thailand. The fruiting body was collected on the base of a livingDipterocarpus tree. -

Wood Decay Fungi in Landscape Trees
Pest Notes, Publication 74109 Revised August 2019 Integrated Pest Management for Home Gardeners and Landscape Professionals Wood Decay Fungi in Landscape Trees everal fungal diseases, sometimes called heart rots, Ssap rots, or canker rots, decay wood in tree trunks Figure 1. White rot of oak. and limbs (Figures 1 and 2). Under conditions favor- ing growth of specific rot fungi, extensive portions of the wood of living trees can decay in a relatively short time (i.e., months to years). Decay fungi reduce wood strength and may kill storage and conductive tissues in the sapwood. While most species of woody plants are subject to trunk and limb decay, older and weaker trees are most susceptible. DAMAGE Decay fungi destroy cell wall components; including cellulose, hemicellulose, and lignin, that make up the woody portion of a tree. Depending on the organism, decay fungi can destroy the living (sapwood) or the central core (heartwood) part of the tree. Decay isn’t always visible on the outside of the tree, except where the bark Figure 2. Heart brown rot in a conifer trunk. has been cut or injured, when a cavity is present, or when rot fungi produce reproductive structures. Wood decay can make trees hazardous, of wood weight can result in 70 to 90% as infected trunks and limbs become loss in wood strength. Many branches unable to support their own weight and that fall from trees appear sound, but fall, especially when stressed by wind, upon analysis, they were colonized by Authors: heavy rain, or other conditions. Decay wood decay organisms. -

Identification, Effects and Management of 5 Types of Decay Organisms Found in Seattle Parks
Identification, Effects and Management of 5 types of decay organisms found in Seattle Parks Chris Rippey, Arborist [email protected] • Third generation Arborist • Grew up in the bay area of California. • Was 16 when I started working with my dad in tree care • I fell in love with tree work, not trees • Managed the preventative tree maintenance programs at Stanford University for 14 years. • Moved to Washington and began working for Seattle Parks 2 ½ years ago Seattle Parks System - 6,412 Total Acres - 4,016 Acres of Developed Park - 2,396 Acres of Natural Area - 480 Parks - >300,000 trees - >16,000 trees in our tree inventory Seward Park 1920 Ravenna Park 1922 What are we focusing on? - 171,615 trees in our Buffer Zone. - Buffer Zone is a 50’ buffer around high use areas like beaches, paved roads and trails, playgrounds…etc) - Buffer Zones are on average 56% of a given park Tree Risk Inspections Terms & Matrix TERM DEFINITION Likelihood of failure and impacts Imminent Failure has started or is most likely to occur in the near future even if there is no weather forces/rare occurrence. Will fail in a storm. Probable Failure may be expected under normal weather within a time frame. Likely to fail in a severe storm. Possible Failure could occur, but is unlikely during normal weather. May fail in a severe storm. Improbable Tree or branch failure not likely under normal conditions and may not fail in severe weather within a time frame. Risk rating High Failed tree or part will likely impact a target. -

Diseases of Trees in the Great Plains
39. Ganoderma Root Rot or White Mottled Rot James T. Blodgett Ganoderma root rot, also called white mottled rot, is caused by the fungus Ganoderma applanatum. This fungus is found in all 50 states and occurs throughout North America and Europe. It is a pathogen and a common wood-decaying fungus of roots and lower stems (butts) of many deciduous and some coniferous trees species. Ganoderma-caused root rot has been reported in live trees, such as apple (Malus spp.), aspen (Populus spp.), basswood (Tilia spp.), beech (Fagus spp.), birch (Betula spp.), cherry (Prunus spp.), citrus (Citrus spp.), cottonwood (Populus spp.), elm (Ulmus spp.), hemlock (Tsuga spp.), hornbeam (Carpinus caroliniana), horsechestnut (Aesculus hippocastanum), black locust (Robinia pseudoacacia) and honeylocust (Gleditsia triacanthos), maple (Acer spp.), mul- berry (Morus spp.), oak (Quercus spp.), spruce (Picea spp.), sycamore (Platanus occidentalis), tulip tree (Liriodendron tulipifera), sweetgum (Liquidambar styraciflua), and willow (Salix spp.). G. applanatum is commonly known as the artist’s conk. The name comes from the use of its fruiting bodies as a drawing medium by artists (fig. 39-1). When the fresh lower surface is rubbed or scratched, it immediately changes Figure 39-1—Drawing on the lower from white to dark brown, producing shading or surface of Ganoderma applanatum conk visible lines. When the conk is dried, drawings (James J. Worrall, U.S. Forest Service). become permanent. Hosts and Distribution In the Great Plains, G. applanatum occurs predominantly in aspen, cottonwoods, and other Populus species. This fungal pathogen is irregularly distributed across the Great Plains, but its distribution is not well documented in many areas. -

Antibacterial Activity, Optimal Culture Conditions and Cultivation of the Medicinal Ganoderma Australe, New to Thailand
1108 Mycosphere 8(8): 1108–1123 (2017) www.mycosphere.org ISSN 2077 7019 Article Doi 10.5943/mycosphere/8/8/11 Copyright © Guizhou Academy of Agricultural Sciences Antibacterial activity, optimal culture conditions and cultivation of the medicinal Ganoderma australe, new to Thailand Luangharn T1,2, Karunarathna SC1,2, Khan S1,2, Xu JC1,2*, Mortimer PE1,2 and Hyde KD1,2,3,4,5 1 Key Laboratory of Economic Plants and Biotechnology, Kunming Institute of Botany, Chinese Academy of Sciences, 132 Lanhei Road, Kunming 650201, China. 2 World Agroforestry Centre, China & East-Asia Office, 132 Lanhei Road, Kunming 650201, China.3 3 Center of Excellence in Fungal Research, Mae Fah Luang University, Chiang Rai 57100, Thailand. 4 School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand. 5 Mushroom Research Foundation, 128 M.3 Ban Pa Deng T. Pa Pae, A. Mae Taeng, Chiang Mai 50150, Thailand. Luangharn T, Karunarathna SC, Khan S, Xu JC, Mortimer PE, Hyde KD 2017 – Antibacterial activity, optimal culture conditions and cultivation of the medicinal Ganoderma australe, new to Thailand. Mycosphere 8(8), 1108–1123, Doi 10.5943/mycosphere/8/8/11 Abstract Ganoderma is a well-known genus of medicinal mushrooms that belongs to the order Polyporales. Many members of this genus are extensively used in traditional Asian medicines. Herein we report a new strain of Ganoderma australe collected in Thailand and identified using macro- and micro-morphological characteristics as well as phylogenetic analysis. The optimal conditions for mycelia growth were 25–30 ºC at pH 7–8, while sorghum and barley were found to be the best grain media for spawn production. -

MUSHROOMS of the OTTAWA NATIONAL FOREST Compiled By
MUSHROOMS OF THE OTTAWA NATIONAL FOREST Compiled by Dana L. Richter, School of Forest Resources and Environmental Science, Michigan Technological University, Houghton, MI for Ottawa National Forest, Ironwood, MI March, 2011 Introduction There are many thousands of fungi in the Ottawa National Forest filling every possible niche imaginable. A remarkable feature of the fungi is that they are ubiquitous! The mushroom is the large spore-producing structure made by certain fungi. Only a relatively small number of all the fungi in the Ottawa forest ecosystem make mushrooms. Some are distinctive and easily identifiable, while others are cryptic and require microscopic and chemical analyses to accurately name. This is a list of some of the most common and obvious mushrooms that can be found in the Ottawa National Forest, including a few that are uncommon or relatively rare. The mushrooms considered here are within the phyla Ascomycetes – the morel and cup fungi, and Basidiomycetes – the toadstool and shelf-like fungi. There are perhaps 2000 to 3000 mushrooms in the Ottawa, and this is simply a guess, since many species have yet to be discovered or named. This number is based on lists of fungi compiled in areas such as the Huron Mountains of northern Michigan (Richter 2008) and in the state of Wisconsin (Parker 2006). The list contains 227 species from several authoritative sources and from the author’s experience teaching, studying and collecting mushrooms in the northern Great Lakes States for the past thirty years. Although comments on edibility of certain species are given, the author neither endorses nor encourages the eating of wild mushrooms except with extreme caution and with the awareness that some mushrooms may cause life-threatening illness or even death. -
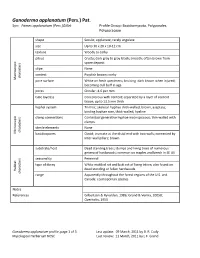
Ganoderma Applanatum (Pers.) Pat
Ganoderma applanatum (Pers.) Pat. Syn: Fomes applanatum (Pers.)Gillet Profile Group: Basidiomycota, Polyporales, Polyporaceae shape Sessile; applanate; rarely ungulate size Up to 30 x 20 x 10-12 cm texture Woody to corky pileus Crusty; dark gray to gray-black; smooth; often brown from spore deposit stipe None context Purplish brown; corky characters Macroscopic pore surface White on fresh specimens; bruising dark brown when injured; becoming dull buff in age pores Circular; 4-6 per mm tube layer(s) Concolorous with context; separated by a layer of context tissue; up to 13.5 mm thick hyphal system Trimitic; skeletal hyphae thick-walled, brown, aseptate; binding hyphae rare, thick-walled, hyaline clamp connections Contextual generative hyphae inconspicuous; thin-walled with clamps sterile elements None characters Microscopic basidiospores Ovoid; truncate at the distal end with two walls; connected by inter-wall pillars; brown substrate/host Dead standing trees; stumps and living trees of numerous genera of hardwoods; common on maples and beech in SE US seasonality Perennial type of decay White mottled rot and butt rot of living trtees; also found on dead standing or fallen hardwoods Habitat characters range Apparently throughout the forest regions of the U.S. and Canada; cosmopolitan species Notes References Gilbertson & Ryvarden, 1986; Grand & Vernia, 2005B; Overholts, 1953 Ganoderma applanatum profile, page 1 of 3 Last update: 09 March, 2011 by B. R. Cody Mycological Herbarium NCSC Last review: 11 March, 2011 by L.F. Grand Species distribution in North Carolina Ganoderma applanatum (Pers.) Pat. Habit of Basidiocarps Habit of Basidiocarps Habit of Basidiocarp Habit of Basidiocarp Ganoderma applanatum profile, page 2 of 3 Last update: 09 March, 2011 by B. -

Field Guide for Danger Tree Identification and Response
United States Department of Agriculture Field Guide for Forest Service Pacific Northwest Danger Tree Region Identification and United States Response Department of Interior Bureau of Land Management Oregon Associated Oregon OSHA Loggers, Inc. Inside Front Cover Field Guide for Danger Tree Identification and Response Richard Toupin Regional Logging Engineer, Gregory Filip Regional Forest Pathologist, Thomas Erkert Transportation Group Leader Pacific Northwest Region USDA Forest Service • Michael Barger Logging Specialist Salem District USDI Bureau of Land Management • 2008 Acknowledgements We thank the following people who contributed to revising this guide: Diane Hildebrand, Donald Goheen, Craig Schmitt, Kristen Chadwick, Angel Saavedra, and Katrina Mallams. Graphic Design: Michael Hamel (FS) The U.S. Department of Agriculture (USDA) prohibits discrimination in all its programs and activities on the basis of race, color, national origin, age, disability, and where applicable, sex, marital status, familial status, parental status, religion, sexual orientation, genetic information, political beliefs, reprisal, or because all or part of an individual’s income is derived from any public assistance program. (Not all prohibited bases apply to all programs.) Persons with disabilities who require alternative means for communication of program information (Braille, large print, audiotape, etc.) should contract USDA’s TARGET Center at 202-720-2600 (voice and TDD). To file a complaint of discrimination, write to USDA, Director, Office of Civil Rights, 1400 Independence Avenue, S.W., Washington, D.C. 20250-9410, or call 800-795-3272 (voice) or 202-720-6382 (TDD). USDA is an equal opportunity provider and employer. R6-NR-FP-PR-01-08 1 2 Table of Contents Introduction ...............................................................5 Regulatory Basis ........................................................6 Responsibilities. -

Biodiversity and Morphological Characterization of Mushrooms at the Tropical Moist Deciduous Forest Region of Bangladesh
American Journal of Experimental Agriculture 8(4): 235-252, 2015, Article no.AJEA.2015.167 ISSN: 2231-0606 SCIENCEDOMAIN international www.sciencedomain.org Biodiversity and Morphological Characterization of Mushrooms at the Tropical Moist Deciduous Forest Region of Bangladesh M. I. Rumainul1, F. M. Aminuzzaman1* and M. S. M. Chowdhury1 1Department of Plant Pathology, Faculty of Agriculture, Sher-e-Bangla Agricultural University, Sher-e-Bangla Nagar, Dhaka-1207, Bangladesh. Authors’ contributions This work was carried out in collaboration with all authors. Author MIR carried out the research and wrote the first draft of the manuscript. Author FMA designed use supervised and edited the manuscript. All authors read and approved the final manuscript. Article Information DOI: 10.9734/AJEA/2015/17301 Editor(s): (1) Sławomir Borek, Department of Plant Physiology, Adam Mickiewicz University, Poland. Reviewers: (1) Anonymous, Ghana. (2) Anonymous, India. (3) Eduardo Bernardi, Departamento de Microbiologia, Universidade Federal de Pelotas, Brazil. Complete Peer review History: http://www.sciencedomain.org/review-history.php?iid=1078&id=2&aid=9182 Received 7th March 2015 th Original Research Article Accepted 17 April 2015 Published 8th May 2015 ABSTRACT Mushroom flora is an important component of the ecosystem and their biodiversity study has been largely neglected and not documented for the tropical moist deciduous forest regions of Bangladesh. This investigation was conducted in seven different areas of tropical moist deciduous forest region of Bangladesh namely Dhaka, Gazipur, Bogra, Rajshahi, Pabna, Jaipurhat and Dinajpur. Mushroom flora associated with these forest regions were collected, photographed and preserved. A total of fifty samples were collected and identified to fourteen genera and twenty four species. -

Common Tree Decays
Evolving Urban Landscapes: Trees, Bees and IPM Strategies, City of Seattle 2018 IPM Seminars Common Tree Decays presented by, Bess Bronstein Horticultural Consultant & ISA Certified Arborist [email protected] Photo: Jay W. Pscheidt, 2018 Plant Pathogenic Fungi Symptoms Susan K. Hagle, USDA Forest Service, Bugwood.org • cankers • galls • leaf spots • leaf blotches • root rots • stem rots • wilts • deformity Signs • mycelia • spores • fruiting bodies Robert L. Anderson, USDA Forest Service, Bugwood.org Fruiting Bodies https://en.wikipedia.org/wiki/Lycoperdon_perlatum puffballs Laura Sims, 2012, PNW Disease Handbook mushrooms conks Magickcanoe.com Fungi Characteristics hypha (pl., hyphae) mycelium (pl., mycelia) • made up of hyphae, which form mycelia • cell wall is mostly chitin, sometimes has cellulose • reproduce by spores (sexual/asexual), budding (asexual), and fragmentation (asexual) Fungi Characteristics http://biology-pictures.blogspot.com Plant Pathogenic Fungi How do they survive? • survival spores • mycelial pieces • fruiting bodies (mushrooms, brackets, conks) • saprophytes in plant debris How do they spread? • infected plant debris • infested seeds • spore dispersal in air • splashing water • tools • insects https://en.wikipedia.org/wiki/Armillaria_mellea Basidiomycete Life Cycle http://www.cals.ncsu.edu Basidiospores Image courtesy of Dr C. Jeffree Wood Decay Fungi white rot: fungus decays cellulose and lignin (decayed wood appears bleached and is often spongy or stringy) brown rot: fungus decays cellulose, but leaves lignin -

Root Rots, Butt Rots & Stem Decays of Alaskan Trees
Root Rots, Butt Rots & Stem Decays of Alaskan Trees DAMAGE AGENTS PAGE Root & Butt Rots (Unknown) 1 Armillaria species / Armillaria Root Rot 3 Heterobasidion occidentale / Heterobasidion Root Rot 5 Onnia tomentosa / Tomentosus Root Rot 7 Phaeolus schweinitzii / Velvet Top 9 Pholiota species / Yellow Cap Fungus 11 Stem Decays (Unknown) 13 Echinodontium tinctorium / Paint Fungus 15 Fomes fomentarius / Tinder Conk 17 Fomitopsis officinalis / Quinine Conk 19 Fomitopsis pinicola / Red Belt Conk 21 Ganoderma applanatum / Artist’s Conk 23 Ganoderma tsugae / Varnish Conk 25 Inonotus obliquus / Cinder Conk 27 Laetiporus sulphureus / Chicken of the woods 29 Phellinus hartigii / Hartigs Conk 31 Phellinus igniarius and P. tremulae/ False Tinder Conk 33 Piptoporus betulinus / Birch Conk 35 Porodaedalea pini / Red Ring Rot 37 0 Root and Butt Rots 1 Root and Butt Rots Hosts: All tree species in Alaska. ID: Decayed roots and butts. Mushrooms, conks, or other fungal structures on the root collar or roots (nonpathogenic mushrooms and conks can also be found near tree bases). Uprooted trees have few remaining roots (root ball, root wad). The direction of tree fall may be inconsistent rather than with the direction of prevailing wind. Remarks: The known root and butt diseases in Alaska are caused by internal wood decay (rot) fungi that may overlap with stem decay fungi. Butt rot fungi decay the heartwood at the base of the stem. Trees can live with butt rot for years or decades. Tree mortality usually occurs due to uprooting or snapping associated with the loss of structural integrity. 2 Armillaria species Armillaria Root Rot mycelial fans 3 rhizomorphs Hosts: All tree species in Alaska.