Histone Methylation by PRC2 Is Inhibited by Active Chromatin Marks
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Four Enzymes Cooperate to Displace Histone H1 During the First Minute of Hormonal Gene Activation
Downloaded from genesdev.cshlp.org on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press Four enzymes cooperate to displace histone H1 during the first minute of hormonal gene activation Guillermo Pablo Vicent,1,2 A. Silvina Nacht,1 Jofre Font-Mateu, Giancarlo Castellano, Laura Gaveglia, Cecilia Ballare´, and Miguel Beato Centre de Regulacio´ Geno` mica (CRG), Universitat Pompeu Fabra (UPF), E-08003 Barcelona, Spain Gene regulation by external signals requires access of transcription factors to DNA sequences of target genes, which is limited by the compaction of DNA in chromatin. Although we have gained insight into how core histones and their modifications influence this process, the role of linker histones remains unclear. Here we show that, within the first minute of progesterone action, a complex cooperation between different enzymes acting on chromatin mediates histone H1 displacement as a requisite for gene induction and cell proliferation. First, activated progesterone receptor (PR) recruits the chromatin remodeling complexes NURF and ASCOM (ASC-2 [activating signal cointegrator-2] complex) to hormone target genes. The trimethylation of histone H3 at Lys 4 by the MLL2/MLL3 subunits of ASCOM, enhanced by the hormone-induced displacement of the H3K4 demethylase KDM5B, stabilizes NURF binding. NURF facilitates the PR-mediated recruitment of Cdk2/CyclinA, which is required for histone H1 displacement. Cooperation of ATP-dependent remodeling, histone methylation, and kinase activation, followed by H1 displacement, is a prerequisite for the subsequent displacement of histone H2A/H2B catalyzed by PCAF and BAF. Chromatin immunoprecipitation (ChIP) and sequencing (ChIP-seq) and expression arrays show that H1 displacement is required for hormone induction of most hormone target genes, some of which are involved in cell proliferation. -

Inferring Causal Relationships Among Different Histone Modifications and Gene Expression
Downloaded from genome.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press Inferring Causal Relationships among Different Histone Modifications and Gene Expression Hong Yu*, Shanshan Zhu*, Bing Zhou*, Huiling Xue and Jing-Dong J. Han Chinese Academy of Sciences Key Laboratory of Molecular Developmental Biology, Center for Molecular Systems Biology, Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Datun Road, Beijing, 100101, China * These authors contributed equally to this work. Correspondence should be addressed to J.-D.J.H ([email protected]). Running title: histone modification and gene expression network Keywords: histone modification, gene expression, Bayesian network, causal relationship, epigenetics 1 Downloaded from genome.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press Abstract Histone modifications are major epigenetic factors regulating gene expression. They play important roles in maintaining stem cell pluripotency and in cancer pathogenesis. Different modifications may combine to form complex ‘histone codes’. Recent high throughput technologies, such as ‘ChIP-chip’ and ‘ChIP-seq’, have generated high resolution maps for many histone modifications on the human genome. Here we use these maps to build a Bayesian network to infer causal and combinatorial relationships among histone modifications and gene expression. A pilot network derived by the same method among polycomb group (PcG) genes and H3K27 trimethylation is accurately supported by current literature. Our unbiased network model among histone modifications is also well supported by cross validation results. It not only confirmed already known relationships, such as those of H3K27me3 to gene silencing, H3K4me3 to gene activation, and the effect of bivalent modification of both H3K4me3 and H3K27me3, but also identified many other relationships that may predict new epigenetic interactions important in epigenetic gene regulation. -

Anti-H3r2me2(Asym) Antibody
FOR RESEARCH USE ONLY! 01/20 Anti-H3R2me2(asym) Antibody CATALOG NO.: A2025-100 (100 µl) BACKGROUND DESCRIPTION: Histones are basic nuclear proteins that are responsible for the nucleosome structure of the chromosomal fiber in eukaryotes. Nucleosomes consist of approximately 146 bp of DNA wrapped around a histone octamer composed of pairs of each of the four core histones (H2A, H2B, H3, and H4). The chromatin fiber is further compacted through the interaction of a linker histone, H1, with the DNA between the nucleosomes to form higher order chromatin structures. This gene is intronless and encodes a replication-dependent histone that is a member of the histone H3 family. Transcripts from this gene lack poly A tails; instead, they contain a palindromic termination element. This gene is located separately from the other H3 genes that are in the histone gene cluster on chromosome 6p22-p21.3. H3.4; H3/g; H3FT; H3t; HIST3H3; Histone H3; HIST1H3A ALTERNATE NAMES: ANTIBODY TYPE: Polyclonal HOST/ISOTYPE: Rabbit / IgG IMMUNOGEN: A synthetic methylated peptide targeting residues around Arginine 2 of human Histone H3 MOLECULAR WEIGHT: 17 kDa PURIFICATION: Affinity purified FORM: Liquid FORMULATION: In PBS with 0.02% sodium azide, 50% glycerol, pH 7.3 SPECIES REACTIVITY: Human, Mouse, Rat STORAGE CONDITIONS: Store at -20ºC. Avoid freeze / thaw cycles APPLICATIONS AND USAGE: WB 1:500 - 1:2000, IHC 1:50 - 1:200, IF 1:50 - 1:200 Note: This information is only intended as a guide. The optimal dilutions must be determined by the user Western blot analysis of H3R2me2(asym) expression in Dot-blot analysis of methylation peptides using Anti- HeLa cells and H3 protein. -

Symmetric-Di-Methyl-Histone H3 (Arg8) Rabbit
Symmetric -Di-methyl-Histone H3 (Arg8) rabbit pAb ( H3R8me2(sy) pAb) www.ptm-biolab.com.cn Cat#: PTM-672 [email protected] Species: Rabbit 0571-2883 3567 Size: 100 μl Form: supplied in liquid form Recommend Species Swissprot Molecular Application Immunogen dilution Reactivity ID Weight H3R8me2(sy) WB, Dot, ELISA 1:2000 for WB H, M, R, Mk P68431 17 KDa -KLH **Species reactivity is determined by WB. Kept at -20oC after reconstituted. *** Anti-rabbit secondary antibodies must be used to detect this antibody. Source/Purification: This product is produced by immunizing rabbits with a synthetic di-methyl(sy) peptide corresponding to residues surrounding Arg8 of human histone H3. Antibodies are purified by protein A-conjugated agarose followed by di-methylated(sy) histone H3 (Arg8) peptide affinity chromatography. Recommended Applications: ELISA, Dot blot, Western blot. Recommended antibody dilution: WB: 1:2000 NOTE: For WB, incubate membrane with diluted antibody in 5% nonfat milk, 1 x TBS, 0.1% Tween- 20 for two hours at room temperature with gentle shaking. Prepare working dilution immediately before use. Use at an assay dependent concentration. Optimal dilutions/concentrations should be determined by the end user. Not yet tested in other applications Scientific Description: Proteins post-translational Modification (PTM) is an important reversible event controlling protein activity. The amino-terminal tails of core histones undergo multiple modifications in multiple sites, termed as “histone code” or “epigenetic code”. Lysine methylation or arginine methylation in core histones is a major determinant for the formation of active and inactive regions of the genome and therefore plays vital roles in multiple cellular events. -

BMC Genomics Biomed Central
BMC Genomics BioMed Central Research article Open Access Determination of enriched histone modifications in non-genic portions of the human genome Jeffrey A Rosenfeld1,2,3, Zhibin Wang4, Dustin E Schones4, Keji Zhao4, Rob DeSalle3 and Michael Q Zhang*1 Address: 1Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 11724 USA, 2Department of Biology, New York University, New York, NY USA, 3American Museum of Natural History, New York, NY USA and 4Laboratory of Molecular Immunology, National Heart, Lung and Blood Institute, NIH, Bethesda, MD USA Email: Jeffrey A Rosenfeld - [email protected]; Zhibin Wang - [email protected]; Dustin E Schones - [email protected]; Keji Zhao - [email protected]; Rob DeSalle - [email protected]; Michael Q Zhang* - [email protected] * Corresponding author Published: 31 March 2009 Received: 8 September 2008 Accepted: 31 March 2009 BMC Genomics 2009, 10:143 doi:10.1186/1471-2164-10-143 This article is available from: http://www.biomedcentral.com/1471-2164/10/143 © 2009 Rosenfeld et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Chromatin immunoprecipitation followed by high-throughput sequencing (ChIP- seq) has recently been used to identify the modification patterns for the methylation and acetylation of many different histone tails in genes and enhancers. Results: We have extended the analysis of histone modifications to gene deserts, pericentromeres and subtelomeres. Using data from human CD4+ T cells, we have found that each of these non- genic regions has a particular profile of histone modifications that distinguish it from the other non- coding regions. -

Le G´Enome En Action
LEGENOME´ EN ACTION SEQUENC´ ¸ AGE HAUT DEBIT´ ET EPIG´ ENOMIQUE´ Epig´enomique´ ? IFT6299 H2014 ? UdeM ? Mikl´osCs}ur¨os Regulation´ d’expression la transcription d’une region´ de l’ADN necessite´ ? liaisons proteine-ADN´ (facteur de transcription et son site reconnu) ? accessibilite´ de la chromatine REVIEWS Identification of regions that control transcription An initial step in the analysis of any gene is the identifi- cation of larger regions that might harbour regulatory control elements. Several advances have facilitated the prediction of such regions in the absence of knowl- edge about the specific characteristics of individual cis- Chromatin regulatory elements. These tools broadly fall into two categories: promoter (transcription start site; TSS) and enhancer detection. The methods are influenced Distal TFBS by sequence conservation between ORTHOLOGOUS genes (PHYLOGENETIC FOOTPRINTING), nucleotide composition and the assessment of available transcript data. Functional regulatory regions that control transcrip- tion rates tend to be proximal to the initiation site(s) of transcription. Although there is some circularity in the Co-activator complex data-collection process (regulatory sequences are sought near TSSs and are therefore found most often in these regions), the current set of laboratory-annotated regula- tory sequences indicates that sequences near a TSS are Transcription more likely to contain functionally important regulatory initiation complex Transcription controls than those that are more distal. However, specifi- initiation cation of the position of a TSS can be difficult. This is fur- ther complicated by the growing number of genes that CRM Proximal TFBS selectively use alternative start sites in certain contexts. Underlying most algorithms for promoter prediction is a Figure 1 | Components of transcriptional regulation. -
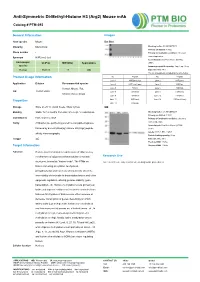
Anti-Symmetric Di-Methyl-Histone H3 (Arg2) Mouse Mab
Anti-Symmetric Di-Methyl-Histone H3 (Arg2) Mouse mAb Catalog # PTM-693 General Information Images Host species Mouse Dot Blot Clonality Monoclonal Blocking buffer: 5% NFDM/TBST Primary ab dilution: 1:1000 Clone number / Primary ab incubation condition: 2 hours at room temperature Synonym H3R2me2 (sy) Secondary ab: Goat Anti-Mouse IgG H&L Immunogen UniProt MW (kDa) Applications (HRP) species Immunogen peptide quantity: 1 ng, 4 ng, 16 ng Human P68431 17 WB Exposure time: 60 s The list of peptides is included in the table below: No. Peptide No. Peptide Product Usage Information Lane 1 H3R2me2 (sy) Lane 2 H3R2me1 Application Dilution Recommended species Lane 3 H3R2me2 (asy) Lane 4 H3K4ac Human, Mouse, Rat, Lane 5 H3K4cr Lane 6 H3K4hib WB 1:500-1:2000 Lane 7 H3K4me1 Lane 8 H3K4me2 Chlorocebus aethiops Lane 9 H3K4me3 Lane 10 H3R8me1 Lane 11 H3R8me2 Lane 12 H3R8me2 (asy) Properties Lane 13 H3R2un Storage Store at -20 °C. Avoid freeze / thaw cycles. WB Blocking buffer: 5% NFDM/TBST Stability Stable for 12 months from date of receipt / reconstitution Primary ab dilution: 1:2000 Constituents PBS, Glycerol, BSA Primary ab incubation condition: 2 hours at room temperature Purity Antibodies are purified by protein G-conjugated agarose Secondary ab: Goat Anti-Mouse IgG H&L followed by di-methylated(sy) histone H3 (Arg2) peptide (HRP) Lysate: MCF-7, BRL, COS-7 affinity chromatography. Protein loading quantity: 20 μg Isotype IgG Exposure time: 60 s Predicted MW: 17 kDa Target Information Observed MW: 17 kDa Function Histone post-translational modifications (PTMs) are key mechanisms of epigenetics that modulate chromatin Research Use structures, termed as “histone code”. -

H3r2me2(Asym) Polyclonal Antibody - Classic
H3R2me2(asym) polyclonal antibody - Classic Cat. No. C15410316 Specificity: Human: positive / Other species: not tested Type: Polyclonal ChIP-grade Purity: Affinity purified polyclonal antibody in PBS containing Source: Rabbit 0.02% azide and 50% glycerol. Lot #: 001 Storage: Store at -20°C; for long storage, store at -80°C Avoid multiple freeze-thaw cycles Size: 50 μg /50 μl Precautions: This product is for research use only Concentration: 1 μg/μl Not for use in diagnostic or therapeutic procedures Description : Polyclonal antibody raised in rabbit against the region of histone H3 containing the asymmetrically dimethylated Arginine 2 (H3R2me2(asym)), using a KLH-conjugated synthetic peptide. Applications Applications Suggested dilution/amount Results ChIP* 2 μg/ChIP Fig 1 Western blotting 1:1,000 Fig 2 * Please note that the optimal antibody amount per IP should be determined by the end-user. We recommend testing 1-5 μl per IP. Target description Histones are the main constituents of the protein part of chromosomes of eukaryotic cells. They are rich in the amino acids arginine and lysine and have been greatly conserved during evolution. Histones pack the DNA into tight masses of chromatin. Two core histones of each class H2A, H2B, H3 and H4 assemble and are wrapped by 146 base pairs of DNA to form one octameric nucleosome. Histone tails undergo numerous post-translational modifications, which either directly or indirectly alter chromatin structure to facilitate transcriptional activation or repression or other nuclear processes. In addition to the genetic code, combinations of the different histone modifications reveal the so-called “histone code”. Histone methylation and demethylation is dynamically regulated by respectively histone methyl transferases and histone demethylases. -

(CHR5 and JMJ15) in Gene Expression Yuan Shen
Functional analysis of Arabidopsis chromatin modification and remodeling regulators (CHR5 and JMJ15) in gene expression Yuan Shen To cite this version: Yuan Shen. Functional analysis of Arabidopsis chromatin modification and remodeling regulators (CHR5 and JMJ15) in gene expression. Agricultural sciences. Université Paris Sud - Paris XI, 2014. English. NNT : 2014PA112093. tel-01124347 HAL Id: tel-01124347 https://tel.archives-ouvertes.fr/tel-01124347 Submitted on 6 Mar 2015 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. UNIVERSITE PARIS-SUD ÉCOLE DOCTORALE : SCIENCES DU VÉGÉTAL Institut de Biologie des Plantes DISCIPLINE : BIOLOGIE THÈSE DE DOCTORAT Soutenance prévue le 28/05/2014 par Yuan SHEN Functional analysis of Arabidopsis chromatin modification and remodeling regulators (CHR5 and JMJ15) in gene expression Composition du jury : Directeur de thèse : Dao-Xiu ZHOU PR Université Paris Sud (IBP, Orsay) Rapporteurs : Martine DEVIC DR CNRS (IRD, Montpellier) Pierre CAROL PR UPMC (Campus JUSSIEU, Paris) Examinateurs : Loïc LEPINIEC DR INRA (IJPB, Versailles) Daniel BOUYER CR CNRS (IBMP, Strasbourg) President : Graham NOCTOR PR Université Paris Sud (IBP, Orsay) ACKNOWLEDGEMENTS This work presented here was done at the Institut de Biologie des Plantes, under the supervision of Prof. -

Moonlighting with WDR5: a Cellular Multitasker
Journal of Clinical Medicine Review Moonlighting with WDR5: A Cellular Multitasker Alissa duPuy Guarnaccia and William Patrick Tansey * Department of Cell and Developmental Biology, Vanderbilt University School of Medicine, Nashville, TN 37232, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-615-322-1993 Received: 19 December 2017; Accepted: 18 January 2018; Published: 30 January 2018 Abstract: WDR5 is a highly conserved WD40 repeat-containing protein that is essential for proper regulation of multiple cellular processes. WDR5 is best characterized as a core scaffolding component of histone methyltransferase complexes, but emerging evidence demonstrates that it does much more, ranging from expanded functions in the nucleus through to controlling the integrity of cell division. The purpose of this review is to describe the current molecular understandings of WDR5, discuss how it participates in diverse cellular processes, and highlight drug discovery efforts around WDR5 that may form the basis of new anti-cancer therapies. Keywords: WDR5; WD40 repeat; epigenetics; transcription; cancer; drug discovery 1. Introduction Increased understanding of the complexity of eukaryotic life has led to growing awareness of the phenomenon of ‘moonlighting’, in which a protein characterized in one context is found to play roles in other, often quite diverse, cellular processes [1]. That proteins defy neat and simple labeling is not surprising, but the mechanisms through which this occurs, and the implications it creates, are often intriguing and profound. This review is focused on WDR5, which has been extensively studied in the context of histone methylation, but more recently shown to be a preeminent cellular multitasker. -

Histone Modification Levels Are Predictive for Gene Expression
Histone modification levels are predictive for gene expression Rosa Karlića,b,1, Ho-Ryun Chunga,1,2, Julia Lasserrea, Kristian Vlahovičekb,c, and Martin Vingrona aMax-Planck-Institut für Molekulare Genetik, Department of Computational Molecular Biology, Ihnestraße 73, 14195 Berlin, Germany; bBioinformatics Group, Division of Biology, Faculty of Science, Zagreb University, Horvatovac 102a, 10000 Zagreb, Croatia; and cDepartment of Informatics, University of Oslo, P.O. Box 1080, Blindern, NO-0316 Oslo, Norway Edited by Robert G. Roeder, The Rockefeller University, New York, NY, and approved January 7, 2010 (received for review August 20, 2009) Histones are frequently decorated with covalent modifications. and are tightly regulated to achieve a precise control of gene ex- These histone modifications are thought to be involved in various pression. The regulatory mechanisms depend on the action of chromatin-dependent processes including transcription. To eluci- transcription factors, which facilitate the recruitment of pol II date the relationship between histone modifications and transcrip- and/or chromatin modifying complexes. Histone modifications tion, we derived quantitative models to predict the expression can therefore be viewed as a read out of the activity of transcrip- level of genes from histone modification levels. We found that his- tion factors. In line with this idea, there are established links be- tone modification levels and gene expression are very well corre- tween the distinct steps in the transcription cycle and some -

Moshkovich Et Al. PADI4 Negatively Regulates Breast Cancer Stem Cells 1
Author Manuscript Published OnlineFirst on April 7, 2020; DOI: 10.1158/0008-5472.CAN-19-3018 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Moshkovich et al. PADI4 negatively regulates breast cancer stem cells Peptidylarginine deiminase IV (PADI4) regulates breast cancer stem cells via a novel tumor cell-autonomous suppressor role by Nellie Moshkovich1, Humberto J. Ochoa1, Binwu Tang1, Howard H. Yang1, Yuan Yang1, Jing Huang1, Maxwell P. Lee1 and Lalage M. Wakefield1 1Laboratory of Cancer Biology and Genetics, National Cancer Institute, Bethesda MD 20892, USA Running title: PADI4 negatively regulates breast cancer stem cells Keywords: PADI4, Cancer stem cells, Breast cancer, Citrullination, Epigenetics Financial support: This work was supported by funding from the Intramural Research Program of the National Cancer Institute, Center for Cancer Research, NIH project ZIA BC 005785. Corresponding author: Lalage M. Wakefield D Phil, ORCID ID# 0000-0003-4124-5250 37 Convent Drive, MSC 4255, National Cancer Institute, Building 37, Room 4032A, Bethesda MD 20892-4255 Tel: 240-760-6808 Email: [email protected] Conflict of interest statement: The authors declare no conflicts of interest. Word count: Abstract: 240 Main text: 5772 (excluding acknowledgments, references and figure legends) Number of Figures: 5 Number of Tables: 1 Number of References: 49 1 Downloaded from cancerres.aacrjournals.org on September 30, 2021. © 2020 American Association for Cancer Research. Author Manuscript Published OnlineFirst on April 7, 2020; DOI: 10.1158/0008-5472.CAN-19-3018 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited.