BMC Genomics Biomed Central
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Four Enzymes Cooperate to Displace Histone H1 During the First Minute of Hormonal Gene Activation
Downloaded from genesdev.cshlp.org on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press Four enzymes cooperate to displace histone H1 during the first minute of hormonal gene activation Guillermo Pablo Vicent,1,2 A. Silvina Nacht,1 Jofre Font-Mateu, Giancarlo Castellano, Laura Gaveglia, Cecilia Ballare´, and Miguel Beato Centre de Regulacio´ Geno` mica (CRG), Universitat Pompeu Fabra (UPF), E-08003 Barcelona, Spain Gene regulation by external signals requires access of transcription factors to DNA sequences of target genes, which is limited by the compaction of DNA in chromatin. Although we have gained insight into how core histones and their modifications influence this process, the role of linker histones remains unclear. Here we show that, within the first minute of progesterone action, a complex cooperation between different enzymes acting on chromatin mediates histone H1 displacement as a requisite for gene induction and cell proliferation. First, activated progesterone receptor (PR) recruits the chromatin remodeling complexes NURF and ASCOM (ASC-2 [activating signal cointegrator-2] complex) to hormone target genes. The trimethylation of histone H3 at Lys 4 by the MLL2/MLL3 subunits of ASCOM, enhanced by the hormone-induced displacement of the H3K4 demethylase KDM5B, stabilizes NURF binding. NURF facilitates the PR-mediated recruitment of Cdk2/CyclinA, which is required for histone H1 displacement. Cooperation of ATP-dependent remodeling, histone methylation, and kinase activation, followed by H1 displacement, is a prerequisite for the subsequent displacement of histone H2A/H2B catalyzed by PCAF and BAF. Chromatin immunoprecipitation (ChIP) and sequencing (ChIP-seq) and expression arrays show that H1 displacement is required for hormone induction of most hormone target genes, some of which are involved in cell proliferation. -

Subcloning, Expression, and Enzymatic Study of PRMT5
Georgia State University ScholarWorks @ Georgia State University Biology Theses Department of Biology Summer 7-12-2010 Subcloning, Expression, and Enzymatic Study of PRMT5 Ran Guo Georgia State University Follow this and additional works at: https://scholarworks.gsu.edu/biology_theses Part of the Biology Commons Recommended Citation Guo, Ran, "Subcloning, Expression, and Enzymatic Study of PRMT5." Thesis, Georgia State University, 2010. https://scholarworks.gsu.edu/biology_theses/26 This Thesis is brought to you for free and open access by the Department of Biology at ScholarWorks @ Georgia State University. It has been accepted for inclusion in Biology Theses by an authorized administrator of ScholarWorks @ Georgia State University. For more information, please contact [email protected]. SUBCLONING, EXPRESSION, AND ENZYMATIC STUDY OF PRMT5 by RAN GUO Under the Direction of Yujun George Zheng ABSTRACT Protein arginine methyltransferases (PRMTs) mediate the transfer of methyl groups to arginine residues in histone and non-histone proteins. PRMT5 is an important member of PRMTs which symmetrically dimethylates arginine 8 in histone H3 (H3R8) and arginine 3 in histone H4 (H4R3). PRMT5 was reported to inhibit some tumor suppressors in leukemia and lymphoma cells and regulate p53 gene, through affecting the promoter of p53. Through methylation of H4R3, PRMT5 can recruit DNA-methyltransferase 3A (DNMT3A) which regulates gene transcription. All the above suggest that PRMT5 has an important function of suppressing cell apoptosis and is a potential anticancer target. Currently, the enzymatic activities of PRMT5 are not clearly understood. In our study, we improved the protein expression methodology and greatly enhanced the yield and quality of the recombinant PRMT5. -

Inferring Causal Relationships Among Different Histone Modifications and Gene Expression
Downloaded from genome.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press Inferring Causal Relationships among Different Histone Modifications and Gene Expression Hong Yu*, Shanshan Zhu*, Bing Zhou*, Huiling Xue and Jing-Dong J. Han Chinese Academy of Sciences Key Laboratory of Molecular Developmental Biology, Center for Molecular Systems Biology, Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Datun Road, Beijing, 100101, China * These authors contributed equally to this work. Correspondence should be addressed to J.-D.J.H ([email protected]). Running title: histone modification and gene expression network Keywords: histone modification, gene expression, Bayesian network, causal relationship, epigenetics 1 Downloaded from genome.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press Abstract Histone modifications are major epigenetic factors regulating gene expression. They play important roles in maintaining stem cell pluripotency and in cancer pathogenesis. Different modifications may combine to form complex ‘histone codes’. Recent high throughput technologies, such as ‘ChIP-chip’ and ‘ChIP-seq’, have generated high resolution maps for many histone modifications on the human genome. Here we use these maps to build a Bayesian network to infer causal and combinatorial relationships among histone modifications and gene expression. A pilot network derived by the same method among polycomb group (PcG) genes and H3K27 trimethylation is accurately supported by current literature. Our unbiased network model among histone modifications is also well supported by cross validation results. It not only confirmed already known relationships, such as those of H3K27me3 to gene silencing, H3K4me3 to gene activation, and the effect of bivalent modification of both H3K4me3 and H3K27me3, but also identified many other relationships that may predict new epigenetic interactions important in epigenetic gene regulation. -

Anti-H3r2me2(Asym) Antibody
FOR RESEARCH USE ONLY! 01/20 Anti-H3R2me2(asym) Antibody CATALOG NO.: A2025-100 (100 µl) BACKGROUND DESCRIPTION: Histones are basic nuclear proteins that are responsible for the nucleosome structure of the chromosomal fiber in eukaryotes. Nucleosomes consist of approximately 146 bp of DNA wrapped around a histone octamer composed of pairs of each of the four core histones (H2A, H2B, H3, and H4). The chromatin fiber is further compacted through the interaction of a linker histone, H1, with the DNA between the nucleosomes to form higher order chromatin structures. This gene is intronless and encodes a replication-dependent histone that is a member of the histone H3 family. Transcripts from this gene lack poly A tails; instead, they contain a palindromic termination element. This gene is located separately from the other H3 genes that are in the histone gene cluster on chromosome 6p22-p21.3. H3.4; H3/g; H3FT; H3t; HIST3H3; Histone H3; HIST1H3A ALTERNATE NAMES: ANTIBODY TYPE: Polyclonal HOST/ISOTYPE: Rabbit / IgG IMMUNOGEN: A synthetic methylated peptide targeting residues around Arginine 2 of human Histone H3 MOLECULAR WEIGHT: 17 kDa PURIFICATION: Affinity purified FORM: Liquid FORMULATION: In PBS with 0.02% sodium azide, 50% glycerol, pH 7.3 SPECIES REACTIVITY: Human, Mouse, Rat STORAGE CONDITIONS: Store at -20ºC. Avoid freeze / thaw cycles APPLICATIONS AND USAGE: WB 1:500 - 1:2000, IHC 1:50 - 1:200, IF 1:50 - 1:200 Note: This information is only intended as a guide. The optimal dilutions must be determined by the user Western blot analysis of H3R2me2(asym) expression in Dot-blot analysis of methylation peptides using Anti- HeLa cells and H3 protein. -

Symmetric-Di-Methyl-Histone H3 (Arg8) Rabbit
Symmetric -Di-methyl-Histone H3 (Arg8) rabbit pAb ( H3R8me2(sy) pAb) www.ptm-biolab.com.cn Cat#: PTM-672 [email protected] Species: Rabbit 0571-2883 3567 Size: 100 μl Form: supplied in liquid form Recommend Species Swissprot Molecular Application Immunogen dilution Reactivity ID Weight H3R8me2(sy) WB, Dot, ELISA 1:2000 for WB H, M, R, Mk P68431 17 KDa -KLH **Species reactivity is determined by WB. Kept at -20oC after reconstituted. *** Anti-rabbit secondary antibodies must be used to detect this antibody. Source/Purification: This product is produced by immunizing rabbits with a synthetic di-methyl(sy) peptide corresponding to residues surrounding Arg8 of human histone H3. Antibodies are purified by protein A-conjugated agarose followed by di-methylated(sy) histone H3 (Arg8) peptide affinity chromatography. Recommended Applications: ELISA, Dot blot, Western blot. Recommended antibody dilution: WB: 1:2000 NOTE: For WB, incubate membrane with diluted antibody in 5% nonfat milk, 1 x TBS, 0.1% Tween- 20 for two hours at room temperature with gentle shaking. Prepare working dilution immediately before use. Use at an assay dependent concentration. Optimal dilutions/concentrations should be determined by the end user. Not yet tested in other applications Scientific Description: Proteins post-translational Modification (PTM) is an important reversible event controlling protein activity. The amino-terminal tails of core histones undergo multiple modifications in multiple sites, termed as “histone code” or “epigenetic code”. Lysine methylation or arginine methylation in core histones is a major determinant for the formation of active and inactive regions of the genome and therefore plays vital roles in multiple cellular events. -

Le G´Enome En Action
LEGENOME´ EN ACTION SEQUENC´ ¸ AGE HAUT DEBIT´ ET EPIG´ ENOMIQUE´ Epig´enomique´ ? IFT6299 H2014 ? UdeM ? Mikl´osCs}ur¨os Regulation´ d’expression la transcription d’une region´ de l’ADN necessite´ ? liaisons proteine-ADN´ (facteur de transcription et son site reconnu) ? accessibilite´ de la chromatine REVIEWS Identification of regions that control transcription An initial step in the analysis of any gene is the identifi- cation of larger regions that might harbour regulatory control elements. Several advances have facilitated the prediction of such regions in the absence of knowl- edge about the specific characteristics of individual cis- Chromatin regulatory elements. These tools broadly fall into two categories: promoter (transcription start site; TSS) and enhancer detection. The methods are influenced Distal TFBS by sequence conservation between ORTHOLOGOUS genes (PHYLOGENETIC FOOTPRINTING), nucleotide composition and the assessment of available transcript data. Functional regulatory regions that control transcrip- tion rates tend to be proximal to the initiation site(s) of transcription. Although there is some circularity in the Co-activator complex data-collection process (regulatory sequences are sought near TSSs and are therefore found most often in these regions), the current set of laboratory-annotated regula- tory sequences indicates that sequences near a TSS are Transcription more likely to contain functionally important regulatory initiation complex Transcription controls than those that are more distal. However, specifi- initiation cation of the position of a TSS can be difficult. This is fur- ther complicated by the growing number of genes that CRM Proximal TFBS selectively use alternative start sites in certain contexts. Underlying most algorithms for promoter prediction is a Figure 1 | Components of transcriptional regulation. -

Histone Methylation by PRC2 Is Inhibited by Active Chromatin Marks
Histone methylation by PRC2 is inhibited by active chromatin marks Inauguraldissertation zur Erlangung der Würde eines Doktors der Philosophie vorgelegt der Philosophisch–Naturwissenschaftlichen Fakultät der Universität Basel von Frank W. Schmitges aus Deutschland Basel, 2013 Genehmigt von der Philosophisch-Naturwissenschaftlichen Fakultät auf Antrag von Prof. Dr. Susan Gasser, Dr. Nicolas Thomä, Dr. Wolfgang Fischle. Basel, den 20. September 2011 Prof. Dr. Susan Gasser (Fakultätsverantwortliche) Prof. Dr. Martin Spiess (Dekan) Table of Contents Abbreviations............................................................................................................................ 6 1. Summary ............................................................................................................................... 8 2. Introduction .......................................................................................................................... 9 2.1 Chromatin ......................................................................................................................... 9 2.2 Epigenetics ........................................................................................................................ 9 2.3 Mechanisms of epigenetic regulation ............................................................................. 10 2.3.1 DNA methylation ..................................................................................................... 11 2.3.2 Histone methylation ................................................................................................ -
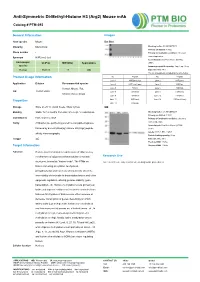
Anti-Symmetric Di-Methyl-Histone H3 (Arg2) Mouse Mab
Anti-Symmetric Di-Methyl-Histone H3 (Arg2) Mouse mAb Catalog # PTM-693 General Information Images Host species Mouse Dot Blot Clonality Monoclonal Blocking buffer: 5% NFDM/TBST Primary ab dilution: 1:1000 Clone number / Primary ab incubation condition: 2 hours at room temperature Synonym H3R2me2 (sy) Secondary ab: Goat Anti-Mouse IgG H&L Immunogen UniProt MW (kDa) Applications (HRP) species Immunogen peptide quantity: 1 ng, 4 ng, 16 ng Human P68431 17 WB Exposure time: 60 s The list of peptides is included in the table below: No. Peptide No. Peptide Product Usage Information Lane 1 H3R2me2 (sy) Lane 2 H3R2me1 Application Dilution Recommended species Lane 3 H3R2me2 (asy) Lane 4 H3K4ac Human, Mouse, Rat, Lane 5 H3K4cr Lane 6 H3K4hib WB 1:500-1:2000 Lane 7 H3K4me1 Lane 8 H3K4me2 Chlorocebus aethiops Lane 9 H3K4me3 Lane 10 H3R8me1 Lane 11 H3R8me2 Lane 12 H3R8me2 (asy) Properties Lane 13 H3R2un Storage Store at -20 °C. Avoid freeze / thaw cycles. WB Blocking buffer: 5% NFDM/TBST Stability Stable for 12 months from date of receipt / reconstitution Primary ab dilution: 1:2000 Constituents PBS, Glycerol, BSA Primary ab incubation condition: 2 hours at room temperature Purity Antibodies are purified by protein G-conjugated agarose Secondary ab: Goat Anti-Mouse IgG H&L followed by di-methylated(sy) histone H3 (Arg2) peptide (HRP) Lysate: MCF-7, BRL, COS-7 affinity chromatography. Protein loading quantity: 20 μg Isotype IgG Exposure time: 60 s Predicted MW: 17 kDa Target Information Observed MW: 17 kDa Function Histone post-translational modifications (PTMs) are key mechanisms of epigenetics that modulate chromatin Research Use structures, termed as “histone code”. -

Histone Methylation Regulation in Neurodegenerative Disorders
International Journal of Molecular Sciences Review Histone Methylation Regulation in Neurodegenerative Disorders Balapal S. Basavarajappa 1,2,3,4,* and Shivakumar Subbanna 1 1 Division of Analytical Psychopharmacology, Nathan Kline Institute for Psychiatric Research, Orangeburg, NY 10962, USA; [email protected] 2 New York State Psychiatric Institute, New York, NY 10032, USA 3 Department of Psychiatry, College of Physicians & Surgeons, Columbia University, New York, NY 10032, USA 4 New York University Langone Medical Center, Department of Psychiatry, New York, NY 10016, USA * Correspondence: [email protected]; Tel.: +1-845-398-3234; Fax: +1-845-398-5451 Abstract: Advances achieved with molecular biology and genomics technologies have permitted investigators to discover epigenetic mechanisms, such as DNA methylation and histone posttransla- tional modifications, which are critical for gene expression in almost all tissues and in brain health and disease. These advances have influenced much interest in understanding the dysregulation of epigenetic mechanisms in neurodegenerative disorders. Although these disorders diverge in their fundamental causes and pathophysiology, several involve the dysregulation of histone methylation- mediated gene expression. Interestingly, epigenetic remodeling via histone methylation in specific brain regions has been suggested to play a critical function in the neurobiology of psychiatric disor- ders, including that related to neurodegenerative diseases. Prominently, epigenetic dysregulation currently brings considerable interest as an essential player in neurodegenerative disorders, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), Amyotrophic lateral sclerosis (ALS) and drugs of abuse, including alcohol abuse disorder, where it may facilitate connections between genetic and environmental risk factors or directly influence disease-specific Citation: Basavarajappa, B.S.; Subbanna, S. -

Stepwise Histone Modifications Are Mediated by Multiple Enzymes That
ARTICLE Received 4 Apr 2013 | Accepted 29 Oct 2013 | Published 26 Nov 2013 DOI: 10.1038/ncomms3841 Stepwise histone modifications are mediated by multiple enzymes that rapidly associate with nascent DNA during replication Svetlana Petruk1, Kathryn L. Black1, Sina K. Kovermann2, Hugh W. Brock2 & Alexander Mazo1 The mechanism of epigenetic inheritance following DNA replication may involve dissociation of chromosomal proteins from parental DNA and reassembly on daughter strands in a specific order. Here we investigated the behaviour of different types of chromosomal proteins using newly developed methods that allow assessment of the assembly of proteins during DNA replication. Unexpectedly, most chromatin-modifying proteins tested, including methylases, demethylases, acetyltransferases and a deacetylase, are found in close proximity to PCNA or associate with short nascent DNA. Histone modifications occur in a temporal order following DNA replication, mediated by complex activities of different enzymes. In contrast, components of several major nucleosome-remodelling complexes are dissociated from parental DNA, and are later recruited to nascent DNA following replication. Epigenetic inheritance of gene expression patterns may require many aspects of chromatin structure to remain in close proximity to the replication complex followed by reassembly on nascent DNA shortly after replication. 1 Department of Biochemistry and Molecular Biology and Kimmel Cancer Center, Thomas Jefferson University, 1020 Locust Street, Philadelphia, Pennsylvania 19107, USA. 2 Department of Zoology, University of British Columbia, 6270 University Boulevard, Vancouver, British Columbia, Canada V6T 1Z4. Correspondence and requests for materials should be addressed to A.M. (email: [email protected]). NATURE COMMUNICATIONS | 4:2841 | DOI: 10.1038/ncomms3841 | www.nature.com/naturecommunications 1 & 2013 Macmillan Publishers Limited. -

Global Histone Modifications in Breast Cancer Correlate with Tumor Phenotypes, Prognostic Factors, and Patient Outcome
Published OnlineFirst April 14, 2009; DOI: 10.1158/0008-5472.CAN-08-3907 Research Article Global Histone Modifications in Breast Cancer Correlate with Tumor Phenotypes, Prognostic Factors, and Patient Outcome Somaia E. Elsheikh,1,7 Andrew R. Green,1 Emad A. Rakha,1 Des G. Powe,1 Rabab A. Ahmed,1,8 Hilary M. Collins,2 Daniele Soria,3 Jonathan M. Garibaldi,3 Claire E. Paish,1 Amr A. Ammar,7 Matthew J. Grainge,4 Graham R. Ball,6 Magdy K. Abdelghany,2,9 Luisa Martinez-Pomares,5 David M. Heery,2 and Ian O. Ellis1 1Department of Histopathology, School of Molecular Medical Sciences, University of Nottingham and Nottingham Universities Hospital Trust, Schools of 2Pharmacy and 3Computer Science, 4Division of Epidemiology and Public Health, and 5School of Molecular Medical Sciences, Institute of Infection, Immunity and Inflammation, University of Nottingham; 6Division of Life Sciences, Nottingham Trent University, Nottingham, United Kingdom; 7Department of Pathology, Faculty of Medicine, Menoufyia University, Menoufyia, Egypt; 8Department of Pathology, Faculty of Medicine, Asuit University, Asuit, Egypt; and 9Department of Pathology, Faculty of Medicine, Suez Canal University, Ismailia, Egypt Abstract Introduction Post-translational histone modifications are known to be Breast cancer is a heterogeneous disease ranging from altered in cancer cells, and loss of selected histone acetylation premalignant hyperproliferation to invasive and metastatic carci- and methylation marks has recently been shown to predict nomas (1). Disease progression is poorly understood but is likely patient outcome in human carcinoma. Immunohistochemistry due to the accumulation of genetic mutations leading to was used to detect a series of histone lysine acetylation widespread changes in gene expression and, in particular, affecting (H3K9ac, H3K18ac, H4K12ac, and H4K16ac), lysine methyla- the expression of tumor suppressors and oncogenes (2). -

H3r2me2(Asym) Polyclonal Antibody - Classic
H3R2me2(asym) polyclonal antibody - Classic Cat. No. C15410316 Specificity: Human: positive / Other species: not tested Type: Polyclonal ChIP-grade Purity: Affinity purified polyclonal antibody in PBS containing Source: Rabbit 0.02% azide and 50% glycerol. Lot #: 001 Storage: Store at -20°C; for long storage, store at -80°C Avoid multiple freeze-thaw cycles Size: 50 μg /50 μl Precautions: This product is for research use only Concentration: 1 μg/μl Not for use in diagnostic or therapeutic procedures Description : Polyclonal antibody raised in rabbit against the region of histone H3 containing the asymmetrically dimethylated Arginine 2 (H3R2me2(asym)), using a KLH-conjugated synthetic peptide. Applications Applications Suggested dilution/amount Results ChIP* 2 μg/ChIP Fig 1 Western blotting 1:1,000 Fig 2 * Please note that the optimal antibody amount per IP should be determined by the end-user. We recommend testing 1-5 μl per IP. Target description Histones are the main constituents of the protein part of chromosomes of eukaryotic cells. They are rich in the amino acids arginine and lysine and have been greatly conserved during evolution. Histones pack the DNA into tight masses of chromatin. Two core histones of each class H2A, H2B, H3 and H4 assemble and are wrapped by 146 base pairs of DNA to form one octameric nucleosome. Histone tails undergo numerous post-translational modifications, which either directly or indirectly alter chromatin structure to facilitate transcriptional activation or repression or other nuclear processes. In addition to the genetic code, combinations of the different histone modifications reveal the so-called “histone code”. Histone methylation and demethylation is dynamically regulated by respectively histone methyl transferases and histone demethylases.