ASMS 2014 Poster Collection
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ranking of Substances for Monitoring in Foods, Drinks and Dietary Supplements - Based on Risk and Knowledge Gaps
VKM Report 2019: 13 Ranking of substances for monitoring in foods, drinks and dietary supplements - based on risk and knowledge gaps Scientific Opinion of the Scientific Steering Committee of the Norwegian Scientific Committee for Food and Environment Scientific Opinion of the Scientific Steering Committee of the Norwegian Scientific Committee for Food and Environment 16.09.2019 ISBN: 978-82-8259-329-8 ISSN: 2535-4019 Norwegian Scientific Committee for Food and Environment (VKM) Po 222 Skøyen N – 0213 Oslo Norway Phone: +47 21 62 28 00 Email: [email protected] vkm.no vkm.no/english Cover photo: Brace Suggested citation: VKM, Inger-Lise Steffensen, Christiane Kruse Fæste, Trine Husøy, Helle Katrine Knutsen, Gro Haarklou Mathisen, Robin Ørnsrud, Angelika Agdestein, Johanna Bodin, Edel Elvevoll, Dag O. Hessen, Merete Hofshagen, Åshild Krogdahl, Asbjørn Magne Nilsen, Trond Rafoss, Taran Skjerdal, Gaute Velle, Yngvild Wasteson, Gro-Ingunn Hemre, Vigdis Vandvik, Jan Alexander (2019). Ranking of substances for monitoring in foods, drinks and dietary supplements - based on risk and knowledge gaps. Scientific Opinion of the Scientific Steering Committee of the Norwegian Scientific Committee for Food and Environment. VKM report 2019:13, ISBN: 978-82-8259-329-8, ISSN: 2535-4019. VKM Report 2019: 13 Ranking of substances for monitoring in foods, drinks and dietary supplements - based on risk and knowledge gaps Preparation of the opinion The Norwegian Scientific Committee for Food and Environment (Vitenskapskomiteen for mat og miljø, VKM) appointed a project group to answer the request from the Norwegian Food Safety Authority. The project group consisted of six VKM members and a project leader from the VKM secretariat. -

Alternaria Alternata Toxins Synergistically Activate the Aryl Hydrocarbon Receptor Pathway in Vitro
biomolecules Article Alternaria alternata Toxins Synergistically Activate the Aryl Hydrocarbon Receptor Pathway In Vitro Julia Hohenbichler 1 , Georg Aichinger 1 , Michael Rychlik 2 , Giorgia Del Favero 1 and Doris Marko 1,* 1 Department of Food Chemistry and Toxicology, University of Vienna, 1090 Vienna, Italy; [email protected] (J.H.); [email protected] (G.A.); [email protected] (G.D.F.) 2 Chair of Analytical Chemistry, Technical University of Munich, 80333 Munich, Germany; [email protected] * Correspondence: [email protected] Received: 18 June 2020; Accepted: 30 June 2020; Published: 9 July 2020 Abstract: Alternaria molds simultaneously produce a large variety of mycotoxins, of which several were previously reported to induce enzymes of phase I metabolism through aryl hydrocarbon receptor activation. Thus, we investigated the potential of naturally occurring Alternaria toxin mixtures to induce Cytochrome P450 (CYP) 1A1/1A2/1B1 activity. Two variants of an extract from cultured Alternaria alternata, as well as the toxins alternariol (AOH), alternariol monomethyl ether (AME), altertoxin I (ATX-I), and altertoxin II (ATX-II), were tested singularly and in binary mixtures applying the 7-ethoxy-resorufin-O-deethylase (EROD) assay in MCF-7 breast cancer cells. Sub-cytotoxic concentrations of the two toxin mixtures, as well as ATX-I, ATX-II and AOH, exhibited dose-dependent enhancements of CYP 1 activity. ATX-I and ATX-II interacted synergistically in this respect, demonstrating the two perylene quinones as major contributors to the extract’s potential. Binary mixtures between AOH and the two altertoxins respectively exhibited concentration-dependent antagonistic as well as synergistic combinatory effects. -

Application of Various Molecular Modelling Methods in the Study of Estrogens and Xenoestrogens
International Journal of Molecular Sciences Review Application of Various Molecular Modelling Methods in the Study of Estrogens and Xenoestrogens Anna Helena Mazurek 1 , Łukasz Szeleszczuk 1,* , Thomas Simonson 2 and Dariusz Maciej Pisklak 1 1 Chair and Department of Physical Pharmacy and Bioanalysis, Department of Physical Chemistry, Medical Faculty of Pharmacy, University of Warsaw, Banacha 1 str., 02-093 Warsaw Poland; [email protected] (A.H.M.); [email protected] (D.M.P.) 2 Laboratoire de Biochimie (CNRS UMR7654), Ecole Polytechnique, 91-120 Palaiseau, France; [email protected] * Correspondence: [email protected]; Tel.: +48-501-255-121 Received: 21 July 2020; Accepted: 1 September 2020; Published: 3 September 2020 Abstract: In this review, applications of various molecular modelling methods in the study of estrogens and xenoestrogens are summarized. Selected biomolecules that are the most commonly chosen as molecular modelling objects in this field are presented. In most of the reviewed works, ligand docking using solely force field methods was performed, employing various molecular targets involved in metabolism and action of estrogens. Other molecular modelling methods such as molecular dynamics and combined quantum mechanics with molecular mechanics have also been successfully used to predict the properties of estrogens and xenoestrogens. Among published works, a great number also focused on the application of different types of quantitative structure–activity relationship (QSAR) analyses to examine estrogen’s structures and activities. Although the interactions between estrogens and xenoestrogens with various proteins are the most commonly studied, other aspects such as penetration of estrogens through lipid bilayers or their ability to adsorb on different materials are also explored using theoretical calculations. -

Natural Products, Synthetic and Non-Nucleoside Compounds As Inhibitors of Enzymes Related to DNA: Update 2013
Send Orders for Reprints to [email protected] Current Enzyme Inhibition, 2014, 10, 13-38 13 Natural Products, Synthetic and Non-Nucleoside Compounds as Inhibitors of Enzymes Related to DNA: Update 2013 Carlos R. Pungitore* INTEQUI-CONICET, Facultad de Química Bioquímicay Farmacia, Universidad Nacional de San Luis, Chacabuco y Pedernera, 5700-San Luis, Argentina Abstract: This review article tries to be an update of 2008 Pungitore’s review. Again, this paper tries to summarize the investigation about natural products, synthetic and non-nucleoside compounds with the ability to inhibit enzymes that play a crucial role in DNA metabolism such as replication, transcription, retro-transcription, recombination, and chromosome segregation during mitosis. The focus is placed on DNA polymerases, topoisomerases and reverse transcriptase inhibitors because most of the literature emphasized on their inhibitory activity. A great diversity of chemical compounds encom- passing triterpenes, flavonoids, chromones, lipids, iridoids, phytosterols, coumarins, anthracyclines, quinones, protober- berines, tannins, lignans, acetogenins, benzimidazoles and other natural products, produced by different species of organ- isms, have inhibitory activities against enzymes related to DNA metabolism allowing these enzymes to arise as important molecular targets for cancer and AIDS research. Keywords: DNA polymerases, inhibitors, natural products, non-nucleoside, reverse transcriptase, topoisomerases. INTRODUCTION There are two classes of RT inhibitors, the nucleoside (NRTIs) and non-nucleoside (NNRTIs) reverse tran- It is well known that natural products play a significant scriptase inhibitors, each disturbing the RT enzyme at a role in drug discovery and development processes. This is different location. NRTIs are the analogs of deoxyribonu- particularly evident for cancer, where over 60% of the drugs cleosides which lack the hydroxyl group on the 3’ carbon currently used in chemotherapy are from natural origin. -

ANSES Report: Infant Total Diet Study (Itds)
Infant Total Diet Study (iTDS) Report 1 ANSES Opinion Summary and conclusions September 2016 Scientific publication Infant Total Diet Study (iTDS) - Report 1 (iTDS) Study Diet Total Infant Infant Total Diet Study (iTDS) Report 1 ANSES Opinion Summary and conclusions September 2016 Scientific publication ANSES Opinion Request No 2014-SA-0317 The Director General Maisons-Alfort, 16 September 2016 OPINION1 of the French Agency for Food, Environmental and Occupational Health & Safety on the dietary exposure of children under three years of age to certain substances ANSES undertakes independent and pluralistic scientific expert assessments. ANSES's public health mission involves ensuring environmental, occupational and food safety as well as assessing the potential health risks they may entail. It also contributes to the protection of the health and welfare of animals, the protection of plant health and the evaluation of the nutritional characteristics of food. It provides the competent authorities with the necessary information concerning these risks as well as the requisite expertise and technical support for drafting legislative and statutory provisions and implementing risk management strategies (Article L.1313-1 of the French Public Health Code). Its opinions are made public. This opinion is a translation of the original French version. In the event of any discrepancy or ambiguity the French language text dated 16 September 2016 shall prevail. On 17 September 2010, the French Agency for Food, Environmental and Occupational Health & Safety issued an internal request for an opinion on the total diet study concerning dietary exposure of children under three years of age to certain substances. In addition to the present opinion, the full study report and a summary are also available on the Agency's website. -

Interaction of Mycotoxin Alternariol with Serum Albumin
International Journal of Molecular Sciences Article Interaction of Mycotoxin Alternariol with Serum Albumin Eszter Fliszár-Nyúl 1,2, Beáta Lemli 2,3,Sándor Kunsági-Máté 2,4, Luca Dellafiora 5 , Chiara Dall’Asta 5, Gabriele Cruciani 6,Gábor Peth˝o 1,7 and Miklós Poór 1,2,* 1 Department of Pharmacology, Faculty of Pharmacy, University of Pécs, Szigeti út 12, H-7642 Pécs; Hungary; [email protected] (E.F.-N.); [email protected] (G.P.) 2 János Szentágothai Research Centre, University of Pécs, Ifjúság útja 20, H-7642 Pécs; Hungary; [email protected] (B.L.); [email protected] (S.K.-M.) 3 Institute of Organic and Medicinal Chemistry, Medical School, University of Pécs, Szigeti út 12, H-7624 Pécs, Hungary 4 Department of Pharmaceutical Chemistry, Faculty of Pharmacy, University of Pécs, Rókus utca 2, H-7642 Pécs, Hungary 5 Department of Food and Drug, University of Parma, Via G.P. 7 Usberti 17/A, 43124 Parma, Italy; luca.dellafi[email protected] (L.D.); [email protected] (C.D.) 6 Department of Chemistry, Biology and Biotechnology, University of Perugia, Via Elce di Sotto 8, 06123 Perugia, Italy; [email protected] 7 Department of Pharmacology and Pharmacotherapy, Medical School, University of Pécs, Szigeti út 12, H-7624 Pécs, Hungary * Correspondence: [email protected]; Tel.: +3672-536-000 (ext. 35052) Received: 19 April 2019; Accepted: 8 May 2019; Published: 12 May 2019 Abstract: Alternariol (AOH) is a mycotoxin produced by Alternaria species. In vitro studies suggest the genotoxic, mutagenic, and endocrine disruptor effects of AOH, and an increased incidence of esophageal cancer has been reported related to higher AOH exposure. -

Current Awareness in Clinical Toxicology Editors: Damian Ballam Msc and Allister Vale MD
Current Awareness in Clinical Toxicology Editors: Damian Ballam MSc and Allister Vale MD August 2015 CONTENTS General Toxicology 6 Metals 34 Management 17 Pesticides 35 Drugs 19 Chemical Warfare 38 Chemical Incidents & 28 Plants 39 Pollution Chemicals 29 Animals 39 CURRENT AWARENESS PAPERS OF THE MONTH Child poisonings with methadone in France: a 6-year prospective national survey since the availability of capsules in 2008 Torrents R, Picot C, Glaizal M, Courne M-A, Schmitt C, Richard N, Simon N, Cardona F, de Haro L. Clin Toxicol 2015; 53: 819-22. Background Methadone for opiate substitution was available only in syrup formulation prior to 2008. In 2007, the French Health Authorities made solid forms available. A national survey was performed in order to evaluate the modification of child poisonings induced by such a new pharmaceutical formulation. Methods A prospective study was set up (April 15, 2008 to April 15, 2014) with the analysis of cases of unintentional ingestion of methadone by patients under 18 years old and managed by the 10 French poison control centers at the national level. As soon as a new pediatric exposure was recorded in the informatics data bank of the Poison Centers, a telephone survey was performed by the Marseilles' Poison Center to obtain the evolution and all the necessary details. Current Awareness in Clinical Toxicology is produced monthly for the American Academy of Clinical Toxicology by the Birmingham Unit of the UK National Poisons Information Service, with contributions from the Cardiff, Edinburgh, and Newcastle Units. The NPIS is commissioned by Public Health England 2 Results 87 cases of child poisonings with the 2 forms were reviewed (syrup, 56 patients; capsules, 31 patients). -

Downloaded 9/29/2021 1:46:45 AM
RSC Advances View Article Online PAPER View Journal | View Issue Potential role of medicinal plants and their constituents in the mitigation of SARS-CoV-2: Cite this: RSC Adv., 2020, 10, 27961 identifying related therapeutic targets using network pharmacology and molecular docking analyses† Eman Shawky, * Ahmed A. Nada and Reham S. Ibrahim Since the outbreak of Coronavirus disease (COVID-19) caused by SARS-CoV-2 in December 2019, there has been no vaccine or specific antiviral medication for treatment of the infection where supportive care and prevention of complications is the current management strategy. In this work, the potential use of medicinal plants and more than 16 500 of their constituents was investigated within two suggested therapeutic strategies in the fight against SARS-CoV-2 including prevention of SARS-CoV-2 RNA Creative Commons Attribution-NonCommercial 3.0 Unported Licence. synthesis and replication, through targeting vital proteins and enzymes as well as modulation of the host's immunity through production of virulence factors. Molecular docking studies on the viral enzymes 3Clpro, PLpro and RdRp suggested rocymosin B, verbascoside, rutin, caftaric acid, luteolin 7-rutinoside, fenugreekine and cyanidin 3-(600-malonylglucoside) as promising molecules for further drug development. Meanwhile, the medicinal plants Glycyrrhiza glabra, Hibiscus sabdariffa, Cichorium intybus, Chrysanthemum coronarium, Nigella sativa, Anastatica hierochuntica, Euphorbia species, Psidium guajava and Epilobium hirsutum were enriched in compounds -

Namenverzeichnis. Author Index
Namenverzeichnis. Author Index Kursiv gedruckte Seitenzahlen beziehen sich auf Literaturverzeichnisse Page numbers printed in italies refer to References Abdel-Kader, M. M. 148 Aota, K. 373 Abdurahman, N. 518 Applegate, H. E. 54 Abe, J. 53 Arakawa, H. 206 Abel, H. 462 Archer, R. A. 53 Abeles, R. H. 624,625,626,627 Ardenne,M.von 394,406,466 Abraham, E. P. 53 Arigoni, D. 458,469,624,628 Abrahamsson, S. 114 Arima, K. 117 Achilladelis, B. 109, 114 Aritomi, M. 187,189,206 Achini, R. 114 Armuth, V. 458 Acker, T. E. 274 Arndt, F. 274 Aczel, St. 216 Arndt, H. G. 210 Adams, M. H. 529,578 Arnold, N. H. 280 Adams, P. M. 114 Aron, E. 274 Adams, R. 219,280 Arroyo, E. R. 408, 448, 458, 466 Adamska, M. 184, 189,206 Arth, G. E. 60 Adinarayana 188 Asao, T. 148 Ager, I. 53 Asen, S. 187, 190 Aichner, F. X. 191,209 Ashbrook, C. W. 57 Alberty, R. A. 623 Asselin, A. 372 Alburn, H. E. 56 Atherton, D. R. 576 Alderweireldt, F. C. 458 Aurnhammer, G. 206,211, 214, 215 Aldrich, P. 518 Algar, J. 199,206 Babel, R. B. 56 Allan, J. 201, 206 Babior, B. M. 623,625 Allen, B. 579 Bader, F. E. 518 Allen, S. H. G. 627 Baeyer, A. 275 Allport, D. C. 274,575 Bagnara, J. D. 575 Alston, R. E. 177, 186,206,212,215 Bailar, J. C. 149 Amos 148 Baird, D. B. 570,575 Andersen, N. H. 283,287,293,304,328,346, Baird, R. -

Occurrence of Alternariol and Alternariol Monomethyl Ether in Apple and Tomato Products and Resistance to Food Processing
Occurrence of alternariol and alternariol monomethyl ether in apple and tomato products and resistance to food processing by (Jason) David Ioi A Thesis presented to The University of Guelph In partial fulfilment of requirements for the degree of Master of Science in Food Science Guelph, Ontario, Canada © (Jason) David Ioi, May, 2017 ABSTRACT OCCURRENCE OF ALTERNARIOL AND ALTERNARIOL MONOMETHYL ETHER IN APPLE AND TOMATO PRODUCTS AND RESISTANCE TO FOOD PROCESSING Jason David Ioi Advisor: University of Guelph, 2017 Professor Massimo F. Marcone The objective of this study was to evaluate the occurrence of alternariol (AOH) and alternariol monomethyl ether (AME) in commercial foods as well as their resistance to food processing techniques. A survey was conducted on the presence of Alternaria mycotoxins in 116 apple and tomato products purchased from Canadian markets in the Spring of 2016. AOH and AME were found in 50% and 30% of these products respectively. The highest concentrations found were 75 µg/kg for AOH and 30 µg/kg for AME. In a second experiment tomato juice spiked with 80, 200, and 500 µg/kg of AOH and AME was subjected to high temperature and high pressure processing. A treatment of 121°C for 20 min reduced AOH content by 13.2% and AME by 15.3%. Processing at 600 MPa for 5 min reduced AOH by 24.9% and 12.8%. ii Acknowledgements I would like to express my deepest gratitude to my advisors for all they have done for me. Thank you for giving me this life changing opportunity. I would like to thank, Dr Massimo Marcone for all of his kindness and guidance in navigating the complexities of academia. -

Role of Dietary Antioxidants
Effect of ochratoxin A on toxicity and gene expression in hepatocytes and kidney tubulus cells – role of dietary antioxidants Dissertation zur Erlangung des Doktorgrades der Mathematisch-Naturwissenschaftlichen Fakultät der Christian-Albrechts-Universität zu Kiel vorgelegt von Christoph Hundhausen Kiel September 2008 MEINEN ELTERN TABLE OF CONTENTS SUMMARY................................................................................................................-I- ZUSAMMENFASSUNG............................................................................................ -II- ABBREVATIONS.....................................................................................................-III - CHAPTER ONE GENERAL INTRODUCTION...............................................................................................................-1- CHAPTER TWO EFFECT OF VITAMIN E ON CYTOCHROME P450 mRNA LEVELS IN CULTURED HEPATOCYTES (HEPG2) AND IN RAT LIVER (PUBLICATION 1)…………………………..….. -14- CHAPTER THREE EFFECT OF VITAMIN E AND POLYPHENOLS ON OCHRATOXIN A-INDUCED CYTOTOXICITY IN LIVER (HEPG2) CELLS (PUBLICATION 2)……………………………….…-23- CHAPTER FOUR OCHRATOXIN A-INDUCED CYTOTOXICITY IN LIVER (HEPG2) CELLS: IMPACT OF SERUM CONCENTRATION, DIETARY ANTIOXIDANTS AND GLUTATHIONE- MODULATING COMPOUNDS (PUBLICATION 3)……………………………………..………….-29- CHAPTER FIVE OCHRATOXIN A IMPAIRS NRF2 DEPENDENT GENE EXPRESSION IN PORCINE KIDNEY TUBULUS CELLS (PUBLICATION 4)………………………………………………..…..-38- CHAPTER SIX OCHRATOXIN A LOWERS mRNA LEVELS OF GENES ENCODING FOR KEY PROTEINS -
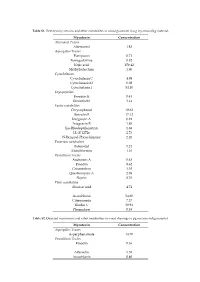
Mycotoxin Concentration Alternaria Toxins Alternariol 1.82 Aspergillus
Table S1. Detected mycotoxins and other metabolites in wood granulate in µg mycotoxin/kg material. Mycotoxin Concentration Alternaria Toxins Alternariol 1.82 Aspergillus Toxins Flavipucin 0.71 Fumigaclavine 0.92 Kojic acid 150.42 Methylsulochrin 1.80 Cytochalasins Cytochalasin C 4.98 Cytochalasin D 0.08 Cytochalasin J 54.40 Depsipeptides Enniatin B 0.41 Enniatin B1 3.14 Exotic metabolites Chrysophanol 19.62 Ilicicolin E 17.12 Integracin A 0.91 Integracin B 1.85 Iso-Rhodoptilometrin 1.88 LL-Z 1272e 2.73 N-Benzoyl-Phenylalanine 2.26 Fusarium metabolites Butenolid 5.24 Moniliformin 1.16 Penicillium Toxins Andrastin A 0.63 Emodin 9.62 Griseofulvin 3.38 Questiomycin A 2.08 Skyrin 8.76 Plant metabolites Abscisic acid 4.72 Ascochlorin 54.60 Citreorosein 7.37 Ilicolin C 30.94 Phomalone 0.38 Table S2. Detected mycotoxins and other metabolites in wood shavings in µg mycotoxin/kg material. Mycotoxin Concentration Aspergillus Toxins Asperphenamate 14.70 Penicillium Toxins Emodin 0.36 Altersetin 5.55 Ascochlorin 0.40 Table S3. Detected mycotoxins and other metabolites in sawdust in µg mycotoxin/kg material. Mycotoxin Concentration Alternaria Toxins Alternariol 10.93 Alternariolmethylether 8.89 Aspergillus Toxins 3-Nitropropionic acid 4.24 Asperphenamate 22.29 Exotic metabolites Iso-Rhodoptilometrin 1.08 LL-Z 1272e 2.73 N-Benzoyl-Phenylalanine 2.26 Fusarium metabolites Aurofusarin 52.81 Monocerin 0.72 Penicillium Toxins Emodin 7.20 Skyrin 3.43 Ascochlorin 6.94 Table S4. Detected mycotoxins and other metabolites in millings in µg mycotoxin/kg material. Mycotoxin Concentration Alternaria Toxins Alternariolmethylether 2.36 Aspergillus Toxins Asperphenamate 3.74 Tryptophol 0.22 Exotic metabolites Ilicicolin A 0.95 Iso-Rhodoptilometrin 0.80 Fusarium metabolites Aurofusarin 40.85 Penicillium Toxins Emodin 2.68 Ascochlorin 9.28 Bassianolide 0.03 Table S5.