Coordinative Binding of Divalent Cations with Ligands Related to Bacterial Spores Equilibrium Studies
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Effects of Calcium Chelation on Digitalis-Induced Cardiac Arrhythmias by Paul Szekely and N
Br Heart J: first published as 10.1136/hrt.25.5.589 on 1 September 1963. Downloaded from Brit. Heart J., 1963, 25, 589. EFFECTS OF CALCIUM CHELATION ON DIGITALIS-INDUCED CARDIAC ARRHYTHMIAS BY PAUL SZEKELY AND N. A. WYNNE From the Cardiovascular Department, Newcastle General Hospital, and the Department ofPhysiology, King's College, University of Durham Received November 19, 1962 Several studies have already shown that cardiac arrhythmias caused by digitalis can be abolished by the induction of hypocalcaemia (Page and Real, 1955; Smith and Grinnell, 1955; Gubner and Kallman, 1957; Kabakow and Brothers, 1958; Surawicz et al., 1959; Cohen et al., 1959; Rosenbaum, Mason, and Seven, 1960; Surawicz, 1960; Soffer, Toribara, and Sayman, 1961). The rationale of inducing hypocalcemia as an anti-arrhythmic measure is based on experimental and clinical obser- vations relating to the direct myocardial action of calcium and to the interaction between calcium, potassium, and digitalis. Low calcium concentration decreases myocardial irritability (Brooks et al., 1955) and it also increases the intracellular potassium concentration (Rosenbaum et al., 1960). There is also a synergistic action between calcium and digitalis, which was observed in the presence of digitalis intoxication (Nalbandian et al., 1957). The present study was undertaken copyright. in order to assess the value of induced hypocalcemia in the management of cardiac arrhythmias caused by digitalis. MATERIAL AND METHODS Forty-eight experiments were carried out under general aneesthesia on 46 cats and 2 dogs. Cardiac arrhythmia was induced by the intravenous administration of tincture of digitalis as previously described http://heart.bmj.com/ (Szekely and Wynne, 1951). -

New Brunswick Drug Plans Formulary
New Brunswick Drug Plans Formulary August 2019 Administered by Medavie Blue Cross on Behalf of the Government of New Brunswick TABLE OF CONTENTS Page Introduction.............................................................................................................................................I New Brunswick Drug Plans....................................................................................................................II Exclusions............................................................................................................................................IV Legend..................................................................................................................................................V Anatomical Therapeutic Chemical (ATC) Classification of Drugs A Alimentary Tract and Metabolism 1 B Blood and Blood Forming Organs 23 C Cardiovascular System 31 D Dermatologicals 81 G Genito Urinary System and Sex Hormones 89 H Systemic Hormonal Preparations excluding Sex Hormones 100 J Antiinfectives for Systemic Use 107 L Antineoplastic and Immunomodulating Agents 129 M Musculo-Skeletal System 147 N Nervous System 156 P Antiparasitic Products, Insecticides and Repellants 223 R Respiratory System 225 S Sensory Organs 234 V Various 240 Appendices I-A Abbreviations of Dosage forms.....................................................................A - 1 I-B Abbreviations of Routes................................................................................A - 4 I-C Abbreviations of Units...................................................................................A -

Chelation Therapy
Medical Policy Chelation Therapy Table of Contents • Policy: Commercial • Coding Information • Information Pertaining to All Policies • Policy: Medicare • Description • References • Authorization Information • Policy History Policy Number: 122 BCBSA Reference Number: 8.01.02 NCD/LCD: N/A Related Policies None Policy Commercial Members: Managed Care (HMO and POS), PPO, and Indemnity Medicare HMO BlueSM and Medicare PPO BlueSM Members Chelation therapy in the treatment of the following conditions is MEDICALLY NECESSARY: • Extreme conditions of metal toxicity • Treatment of chronic iron overload due to blood transfusions (transfusional hemosiderosis) or due to nontransfusion-dependent thalassemia (NTDT) • Wilson's disease (hepatolenticular degeneration), or • Lead poisoning. Chelation therapy in the treatment of the following conditions is MEDICALLY NECESSARY if other modalities have failed: • Control of ventricular arrhythmias or heart block associated with digitalis toxicity • Emergency treatment of hypercalcemia. NaEDTA as chelation therapy is considered NOT MEDICALLY NECESSARY. Off-label applications of chelation therapy are considered INVESTIGATIONAL, including, but not limited to: • Alzheimer’s disease • Arthritis (includes rheumatoid arthritis) • Atherosclerosis, (e.g., coronary artery disease, secondary prevention in patients with myocardial infarction, or peripheral vascular disease) • Autism • Diabetes • Multiple sclerosis. 1 Prior Authorization Information Inpatient • For services described in this policy, precertification/preauthorization IS REQUIRED for all products if the procedure is performed inpatient. Outpatient • For services described in this policy, see below for products where prior authorization might be required if the procedure is performed outpatient. Outpatient Commercial Managed Care (HMO and POS) Prior authorization is not required. Commercial PPO and Indemnity Prior authorization is not required. Medicare HMO BlueSM Prior authorization is not required. -

Post-Injury Calcium Chelation Rescues Skeletal Muscle Regeneration in Mice Matthew .D Magda University of Connecticut - Storrs, [email protected]
University of Connecticut OpenCommons@UConn Honors Scholar Theses Honors Scholar Program Summer 8-1-2013 Post-Injury Calcium Chelation Rescues Skeletal Muscle Regeneration in Mice Matthew .D Magda University of Connecticut - Storrs, [email protected] Follow this and additional works at: https://opencommons.uconn.edu/srhonors_theses Part of the Cell Biology Commons, and the Molecular Biology Commons Recommended Citation Magda, Matthew D., "Post-Injury Calcium Chelation Rescues Skeletal Muscle Regeneration in Mice" (2013). Honors Scholar Theses. 321. https://opencommons.uconn.edu/srhonors_theses/321 1 Post-Injury Calcium Chelation Rescues Skeletal Muscle Regeneration in Mice Honors Thesis Matthew Magda Research Advisor: Dr. Morgan Carlson Honors Advisor: Dr. Kenneth Noll August 2013 2 Abstract: Antibiotics, surgery and organ transplants have pushed average lifespans towards the upper limits of the human body. Drastically reduced morbidity from infection, toxins and traumatic injury have allowed ever greater portions of the populace can reach eighty or ninety years old before dying of old age. Despite the increased role of aging as a source of morbidity, many aspects of aging are poorly characterized. Sarcopenia, progressive muscle loss, and loss of adult myogenic potential, the ability to produce new muscle tissue from adult stem cell sources, are key causes of decreased mobility and strength in aged individuals. If more youthful muscle quality could be restored in old patients they would experience greatly improved quality of life and perhaps even longer lifespans. Satellite cell populations are known to decline sharply by 6-7th decade of life but traditional treatments for sarcopenia, namely exercise intervention, have been shown to exacerbate the degeneration in aged such patients. -

Chelation Therapy
Corporate Medical Policy Chelation Therapy File Name: chelation_therapy Origination: 12/1995 Last CAP Review: 2/2021 Next CAP Review: 2/2022 Last Review: 2/2021 Description of Procedure or Service Chelation therapy is an established treatment for the removal of metal toxins by converting them to a chemically inert form that can be excreted in the urine. Chelation therapy comprises intravenous or oral administration of chelating agents that remove metal ions such as lead, aluminum, mercury, arsenic, zinc, iron, copper, and calcium from the body. Specific chelating agents are used for particular heavy metal toxicities. For example, desferroxamine (not Food and Drug Administration [FDA] approved) is used for patients with iron toxicity, and calcium-ethylenediaminetetraacetic acid (EDTA) is used for patients with lead poisoning. Note that disodium-EDTA is not recommended for acute lead poisoning due to the increased risk of death from hypocalcemia. Another class of chelating agents, called metal protein attenuating compounds (MPACs), is under investigation for the treatment of Alzheimer’s disease, which is associated with the disequilibrium of cerebral metals. Unlike traditional systemic chelators that bind and remove metals from tissues systemically, MPACs have subtle effects on metal homeostasis and abnormal metal interactions. In animal models of Alzheimer’s disease, they promote the solubilization and clearance of β-amyloid protein by binding to its metal-ion complex and also inhibit redox reactions that generate neurotoxic free radicals. MPACs therefore interrupt two putative pathogenic processes of Alzheimer’s disease. However, no MPACs have received FDA approval for treating Alzheimer’s disease. Chelation therapy has also been investigated as a treatment for other indications including atherosclerosis and autism spectrum disorder. -

Chelation of Actinides
UC Berkeley UC Berkeley Previously Published Works Title Chelation of Actinides Permalink https://escholarship.org/uc/item/4b57t174 Author Abergel, RJ Publication Date 2017 DOI 10.1039/9781782623892-00183 Peer reviewed eScholarship.org Powered by the California Digital Library University of California Chapter 6 Chelation of Actinides rebecca J. abergela aChemical Sciences Division, lawrence berkeley National laboratory, One Cyclotron road, berkeley, Ca 94720, USa *e-mail: [email protected] 6.1 The Medical and Public Health Relevance of Actinide Chelation the use of actinides in the civilian industry and defense sectors over the past 60 years has resulted in persistent environmental and health issues, since a large inventory of radionuclides, including actinides such as thorium (th), uranium (U), neptunium (Np), plutonium (pu), americium (am) and curium 1 Downloaded by Lawrence Berkeley National Laboratory on 22/06/2018 20:28:11. (Cm), are generated and released during these activities. Controlled process- Published on 18 October 2016 http://pubs.rsc.org | doi:10.1039/9781782623892-00183 ing and disposal of wastes from the nuclear fuel cycle are the main source of actinide dissemination. however, significant quantities of these radionu- clides have also been dispersed as a consequence of nuclear weapons testing, nuclear power plant accidents, and compromised storage of nuclear materi- als.1 In addition, events of the last fifteen years have heightened public con- cern that actinides may be released as the result of the potential terrorist use of radiological dispersal devices or after a natural disaster affecting nuclear power plants or nuclear material storage sites.2,3 all isotopes of the 15 ele- ments of the actinide series (atomic numbers 89 through 103, Figure 6.1) are radioactive and have the potential to be harmful; the heaviest members, however, are too unstable to be isolated in quantities larger than a few atoms at a time,4 and those elements cited above (U, Np, pu, am, Cm) are the most RSC Metallobiology Series No. -

Effect of Alpha Lipoic Acid on the Blood Cell Count and Iron Kinetics In
Nutr Hosp. 2015;31(2):883-889 ISSN 0212-1611 • CODEN NUHOEQ S.V.R. 318 Original / Farmacia Effect of alpha lipoic acid on the blood cell count and iron kinetics in hypertensive patients Paula Renata Florêncio Mendes, Danielle dos Santos Félix, Paulo César Dantas da Silva, Guêdijany Henrique Pereira and Mônica Oliveira da Silva Simões Dean of Graduate Studies and Research, State University of Paraiba, Campina Grande, Brazil. Abstract EFECTO DEL ÁCIDO LIPOICO EN EL RECUENTO DE SANGRE Y EL METABOLISMO Introduction: The α-lipoic acid (ALA) has been used DE HIERRO EN PACIENTES HIPERTENSOS as a treatment to reduce oxidative damage in Systemic Arterial Hypertension (SAH), but there are no in vivo studies reporting the effect of its mechanism of action on Resumen iron metabolism. Introducción: El Ácido α-Lipóico (ALA) ha sido uti- α Objective: To evaluate the antioxidant effect of -lipoic lizado como recurso terapéutico para reducir daño oxi- acid on Blood cell count (CBC) and iron metabolism in dativo en la Hipertensión Arterial Sistémica (HAS), pero hypertensive subjects with or without anemia. aún no existen estudios in vivo que reporten sobre su me- Method: Double-blind, randomized, placebo-contro- canismo de acción en el metabolismo del hierro. lled clinical trial. The sample consisted of 60 hyperten- Objetivo: Evaluar el efecto antioxidante del ácido Al- sive patients that were randomly divided into treatment fa-Lipóico sobre el hemograma y metabolismo del hierro group (n = 32), receiving 600 mg / day of ALA for twelve en individuos hipertensos con o sin anemia. weeks and control group (n = 28), receiving placebo for Métodos: Estudio clínico doble-ciego, randomizado y the same period. -
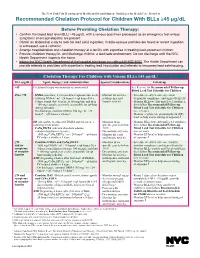
Recommended Chelation Protocol for Children with Blls ≥45 Μg/Dl
The New York City Department of Health and Mental Hygiene Guidelines for Health Care Providers Recommended Chelation Protocol for Children With BLLs ≥45 μg/dL Before Providing Chelation Therapy: • Confirm the blood lead level (BLL) ≥45 μg/dL with a venous specimen processed as an emergency test unless symptoms of encephalopathy are present. • Obtain an abdominal x-ray to look for lead solid ingestion; if radio-opaque particles are found or recent ingestion is witnessed, use a cathartic. • Arrange hospitalization and chelation therapy at a facility with expertise in treating lead-poisoned children. • Provide chelation therapy in, and discharge child to, a lead-safe environment. Do not discharge until the NYC Health Department inspects the home. • Inform the NYC Health Department of the hospital admission by calling 646-632-6002. The Health Department can provide referrals to providers with expertise in treating lead intoxication and referrals to temporary lead-safe housing. Chelation Therapy For Children with Venous BLLs ≥45 μg/dL1 BLL (μg/dL) Agent, Dosage,* and Administration Special Considerations Follow-up <45 Chelation therapy not routinely recommended See Reverse for Recommended Follow-up Blood Lead Test Schedule for Children 45 to <70 • DMSA (succimer, 2,3-meso-dimercaptosuccinic acid) • Monitor for anemia, • Schedule weekly health care visits • 1050 mg DMSA / m2 / 24 hours* ÷ q8 hours PO x neutropenia, and to monitor compliance and signs of toxicity. 5 days; round dose to nearest 100 mg/day, and then hepatic toxicity. • Monitor BLLs weekly until level stabilizes, ÷ 100-mg capsules as evenly as possible for q8-hour then follow Recommended Follow-up dosing schedule. -

Detox for Life: Part 7
Phase III: Cellular Membranes and less accessible matrix detox Objectives y Detox strategies for Phase III y Detox (oral) chelators y Discussion y At home strategies using the various chelators y Handouts: y E-Book y Phase III y Dosing for chlorella and cilantro Phase III: Cellular Membranes and less accessible matrix detox y Detox deposits in extra cellular connective tissues that are more tightly bound to proteins and binding sites on the cells y Detox heavy metal deposits in extra cellular connective tissues that are fibroses, mineralized (plaques in blood vessels) and inaccessible due to hyper-coagulation; y Cellular membranes: this phase starts the intracellular detox, because as the mercury is stripped away from the outside of the cell the intracellular detox mechanisms can transport more mercury to the outside of the cells Phase III: Cellular Membranes and less accessible matrix detox y Keep cellular barriers closed, start to open the brain barrier, but do not aggressively detox the brain yet. y Continue drainage and organ/structure rehabilitation (outlined in Phase II) through detoxification, normalizing blood flow and regulation signaling. y Support the therapeutics of chronic infections that may occur y This is the phase to start adding cilantro to your at home strategies. y Start to rebuild the intracellular stores of glutathione At home: Detox chelators y Chlorella These products are highly considered in Phase III because they penetrate deeper. y Clatherating agents – nano peptides of chlorella and other agents y OSR y Phospho lipids with EDTA y Zeolites Detox chelators Clatherating agents are the nano peptides or the effective binding proteins in chlorella, therefore processed: can be started in phase II y Clatherating agents can be used if chlorella cannot be tolerated y Because clatherating agents are nano particles they are absorbed and more deeply penetrate into the connective tissues. -

Rational Design of Sequestering Agents for Plutonium and Other Actinides
Chem. Rev. 2003, 103, 4207−4282 4207 Rational Design of Sequestering Agents for Plutonium and Other Actinides Anne E. V. Gorden, Jide Xu, and Kenneth N. Raymond* Department of Chemistry, University of California, Berkeley, California 94720 Patricia Durbin Chemical Sciences Division, Ernest Orlando Lawrence Berkeley National Laboratory, University of California, Berkeley, California 94720 Received December 23, 2002 Contents 6.6. Hydroxypyridinone (HOPO)-Based 4246 Plutonium-Sequestering Agents 1. Introduction 4208 6.7. Mixed Ligands 4249 2. Background 4209 6.8. Orally Effective HOPO Ligands 4251 2.1. Actinides in Applications 4209 6.9. Ligand Toxicity 4252 2.2. Behavior of Plutonium in Biological Systems 4209 6.10. Comparison of Efficacy for Pu Sequestration 4254 2.3. Chelation Therapy 4211 6.11. Ligands for Plutonium Removal from Bone 4255 2.4. Current Chelation Methods Using DTPA 4212 7. Thorium-Sequestering Agents 4256 3. Designing a Model System 4213 7.1. Thorium Coordination ChemistrysSimilarities 4256 3.1. Actinide Coordination Chemistry 4213 to Pu(IV) 3.1.1. Characteristic Features 4213 7.2. Structural Characterization as Model for the 4256 3.1.2. Lanthanides as Models for Actinides 4215 Pu(IV) System 3.2. Requirements for an Effective Sequestering 4216 7.3. Decorporation of Thorium 4259 Agent 8. Americium-Sequestering Agents 4260 3.3. Biological Means for Evaluation 4216 8.1. Americium Coordination Chemistry 4260 3.3.1. Initial Assessment 4216 8.2. Lanthanides(III) as Models for Am(III): Eu(III) 4261 3.3.2. Collaborative Studies 4218 and Lu(III) 4. A Biomimetic Approach toward 4219 8.3. Gd(III) Complexes as Models for Am(III) 4262 Actinide-Sequestering Agents 8.4. -

Chelation Therapy
Arizona State Board of Homeopathic Medical Examiners Substantive Policy Statement Regarding Chelation Therapy SPS 04-01 This substantive policy statement is advisory only. A substantive policy statement does not include internal procedural documents that only affect the internal procedures of the agency and does not impose additional requirements or penalties on regulated parties or include confidential information or rules made in accordance with the Arizona administrative procedure act. If you believe that this substantive policy statement does impost additional requirements or penalties on regulated parties you may petition the agency under Arizona Revised Statutes § 41-1033 for a review of the statement. Whereas in A.R.S. § 32-2901 Definitions, states that chelation therapy means an experimental medical therapy to restore cellular homeostasis employing some form of the chelator EDTA (Ethylenediamine Tetraacetic Acid). Chelation therapy is not an experimental therapy if it is used to treat heavy metal poisoning. Therefore, the Board of Homeopathic Medical Examiners’ updated policy statement regarding newer methods of chelation therapy is hereby stated to be the following: It has long been known that the primary function of Chelation or Metal Binding therapy is to deal with Heavy Metal Detoxification. Licensed Homeopathic physicians also utilize chelation therapy in the State of Arizona for purposes such as vascular disease and other non heavy-metal related health problems. There are many different ways to accomplish this goal including the use of several different prescription as well as natural substances that are well known to act as heavy metal chelators. These include pharmacological chelators, such as EDTA, Penicillamine, DMPS, DMSA, Desferoxamine, BAL, as well as many natural chelators and or metal binding substances, such as Ascorbic Acid (Vitamin C), Malic Acid, Garlic, Selenium, and various Amino Acids, etc. -

Heavy Metal Chelation and Testing
Heavy Metal Chelation and Testing Background Information Chelation is the use of a chemical substance to bind molecules, such as metals or minerals, and hold them tightly so they can be removed from the body. The main heavy metal found on testing in the general population is mercury. Fish consumption is directly associated with increased methylmercury levels1, while mercury in dental amalgams is probably the major source of inorganic mercury exposure in humans and studies have shown a direct correlation between the number of amalgam fillings and mercury concentration in blood and urine2. The impact of chronic, low level mercury exposure is now known to adversely impact numerous cellular and organ system processes3. Ionic mercury is antigenic and may contribute significantly to autoimmune processes4, 5. Mercury is also immunotoxic and it may result in immune suppression and allergy6,7,8,9. Research has also demonstrated that multiple strains of antibiotic resistant bacteria develop rapidly in the gut and oral cavity of both humans as well as non-human primates following the placement of amalgam fillings10. Liver glutathione production can be markedly inhibited when mercury accumulates within hepatocytes (liver cells), causing mercury and numerous other toxic substances to accumulate throughout the body11. Mercury may also promote atherosclerosis and hence increase the risk of myocardial infarction12. Methylmercury is considered the most toxic form of mercury and targets the central nervous system whereas inorganic mercury favours accumulation in the kidneys. Research has demonstrated that DMPS13 (2,3-dimercapto-1-propane sulfonic acid) is a good chelating agent for inorganic mercury and DMSA (2,3- dimercaptosuccinic acid ) is considered the most efficient chelator of methylmercury14 and lead15 16.