G-Protein-Coupled Receptor Signaling and Polarized Actin Dynamics 2 Drive Cell-In-Cell Invasion
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

G Protein Alpha 13 (GNA13) (NM 006572) Human Tagged ORF Clone Lentiviral Particle Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC207762L3V G protein alpha 13 (GNA13) (NM_006572) Human Tagged ORF Clone Lentiviral Particle Product data: Product Type: Lentiviral Particles Product Name: G protein alpha 13 (GNA13) (NM_006572) Human Tagged ORF Clone Lentiviral Particle Symbol: GNA13 Synonyms: G13 Vector: pLenti-C-Myc-DDK-P2A-Puro (PS100092) ACCN: NM_006572 ORF Size: 1131 bp ORF Nucleotide The ORF insert of this clone is exactly the same as(RC207762). Sequence: OTI Disclaimer: The molecular sequence of this clone aligns with the gene accession number as a point of reference only. However, individual transcript sequences of the same gene can differ through naturally occurring variations (e.g. polymorphisms), each with its own valid existence. This clone is substantially in agreement with the reference, but a complete review of all prevailing variants is recommended prior to use. More info OTI Annotation: This clone was engineered to express the complete ORF with an expression tag. Expression varies depending on the nature of the gene. RefSeq: NM_006572.3 RefSeq Size: 4744 bp RefSeq ORF: 1134 bp Locus ID: 10672 UniProt ID: Q14344, A0A024R8M0 Domains: G-alpha Protein Families: Druggable Genome Protein Pathways: Long-term depression, Regulation of actin cytoskeleton, Vascular smooth muscle contraction MW: 44 kDa This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 G protein alpha 13 (GNA13) (NM_006572) Human Tagged ORF Clone Lentiviral Particle – RC207762L3V Gene Summary: Guanine nucleotide-binding proteins (G proteins) are involved as modulators or transducers in various transmembrane signaling systems (PubMed:15240885, PubMed:16787920, PubMed:16705036, PubMed:27084452). -

Entosis Enables a Population Response to Starvation
www.impactjournals.com/oncotarget/ Oncotarget, 2017, Vol. 8, (No. 35), pp: 57934-57935 Editorial Entosis enables a population response to starvation Jens C. Hamann and Michael Overholtzer Commentary on: Hamann et al. Entosis is induced by glucose starvation. Cell Rep. 2017; 20:201-210. https://doi.org/10.1016/j. celrep.2017.06.037 Cell death isn’t just about apoptosis anymore. In cells in a population to scavenge extracellular nutrients and the last decade, numerous alternative mechanisms have accumulate the biomass necessary to support proliferation emerged, including regulated forms of necrosis, as well (Figure 1). Macropinocytosis has been shown to act as entosis, a unique mechanism that is executed as a cell similarly to allow cancer cells to scavenge extracellular murder rather than cell suicide [1]. Hamann et al. now proteins [5]. Entosis instead supplies bulk nutrients in identify glucose starvation as a key inducer of this non- the form of whole cells, where, on average, winner cells cell-autonomous form of cell death [2]. ingest two losers, providing a large nutrient supply that Cells that undergo entosis are killed as a result of is well suited to support the outgrowth of selected cells ingestion into their neighbors. They are not phagocytosed, in the population. Indeed, winners with ingested losers but rather form junctions with their neighbors and then have a nearly 10-fold proliferative advantage, and entosis invade into them, ultimately becoming killed through a is required for population re-growth during long-term mechanism involving autophagy proteins and lysosomal starvation. enzymes [1, 3]. This is a competitive process that results Our findings identify entosis as a population-scale in elimination of stiffer cells (“losers”) by softer ones starvation response with parallels to cell competition (“winners”), an effect linked to control over entotic cell occurring in developing tissues [6]. -

PLXNB1 (Plexin
Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL AT INIST-CNRS Gene Section Mini Review PLXNB1 (plexin B1) José Javier Gómez-Román, Montserrat Nicolas Martínez, Servando Lazuén Fernández, José Fernando Val-Bernal Department of Anatomical Pathology, Marques de Valdecilla University Hospital, Medical Faculty, University of Cantabria, Santander, Spain (JJGR, MN, SL, JFVB) Published in Atlas Database: March 2009 Online updated version: http://AtlasGeneticsOncology.org/Genes/PLXNB1ID43413ch3p21.html DOI: 10.4267/2042/44702 This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2010 Atlas of Genetics and Cytogenetics in Oncology and Haematology Identity Pseudogene No. Other names: KIAA0407; MGC149167; OTTHUMP00000164806; PLEXIN-B1; PLXN5; SEP Protein HGNC (Hugo): PLXNB1 Location: 3p21.31 Description Local order: The Plexin B1 gene is located between 2135 Amino acids (AA). Plexins are receptors for axon FBXW12 and CCDC51 genes. molecular guidance molecules semaphorins. Plexin signalling is important in pathfinding and patterning of both neurons and developing blood vessels. Plexin-B1 is a surface cell receptor. When it binds to its ligand SEMA4D it activates several pathways by binding of cytoplasmic ligands, like RHOA activation and subsequent changes of the actin cytoskeleton, axon guidance, invasive growth and cell migration. It monomers and heterodimers with PLXNB2 after proteolytic processing. Binds RAC1 that has been activated by GTP binding. It binds PLXNA1 and by similarity ARHGEF11, Note ARHGEF12, ERBB2, MET, MST1R, RND1, NRP1 Size: 26,200 bases. and NRP2. Orientation: minus strand. This family features the C-terminal regions of various plexins. The cytoplasmic region, which has been called DNA/RNA a SEX domain in some members of this family is involved in downstream signalling pathways, by Description interaction with proteins such as Rac1, RhoD, Rnd1 and other plexins. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

ARHGEF12 Regulates Erythropoiesis and Is Involved in Erythroid Regeneration After Ferrata Storti Foundation Chemotherapy in Acute Lymphoblastic Leukemia Patients
Red Cell Biology & its Disorders ARTICLE ARHGEF12 regulates erythropoiesis and is involved in erythroid regeneration after Ferrata Storti Foundation chemotherapy in acute lymphoblastic leukemia patients Yangyang Xie,1* Li Gao,2* Chunhui Xu,3 Liming Chu,3,7 Lei Gao,4 Ruichi Wu,1 Yu Liu,1 Ting Liu,1 Xiao-jian Sun,5 Ruibao Ren,5 Jingyan Tang,1 Yi Zheng,6 Yong Zhou7 and Shuhong Shen1 1Key Lab of Pediatrics Hematology/Oncology, Ministry of Health, Department of Haematologica 2020 Hematology/Oncology, Shanghai Children's Medical Center, Shanghai Jiao Tong University, Volume 105(4):925-936 Shanghai, China; 2Department of Hematology and Oncology, Children's Hospital of Soochow University, Suzhou, China; 3Shanghai Institute of Nutrition and Health, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Shanghai, China; 4CAS Key Laboratory of Genome Sciences and Information, Beijing Institute of Genomics, Beijing, China; 5State Key Laboratory for Medical Genomics, Shanghai Institute of Hematology, Ruijin Hospital, Shanghai, China; 6Division of Experimental Hematology and Cancer Biology, Cincinnati Children's Hospital Research Foundation, Cincinnati, OH, USA and 7CAS Key Laboratory of Tissue Microenvironment and Tumor, Shanghai Institute of Nutrition and Health, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Shanghai, China *YX and LG contributed equally to this work. ABSTRACT ematopoiesis is a finely regulated process in vertebrates under both homeostatic and stress conditions. By whole exome sequencing, we Correspondence: Hstudied the genomics of acute lymphoblastic leukemia (ALL) YI ZHENG patients who needed multiple red blood cell (RBC) transfusions after inten- [email protected] sive chemotherapy treatment. ARHGEF12, encoding a RhoA guanine YONG ZHOU nucleotide exchange factor, was found to be associated with chemothera- [email protected] py-induced anemia by genome-wide association study analyses. -

G-Protein-Coupled Receptor Signaling and Polarized Actin Dynamics Drive
RESEARCH ARTICLE elifesciences.org G-protein-coupled receptor signaling and polarized actin dynamics drive cell-in-cell invasion Vladimir Purvanov, Manuel Holst, Jameel Khan, Christian Baarlink, Robert Grosse* Institute of Pharmacology, University of Marburg, Marburg, Germany Abstract Homotypic or entotic cell-in-cell invasion is an integrin-independent process observed in carcinoma cells exposed during conditions of low adhesion such as in exudates of malignant disease. Although active cell-in-cell invasion depends on RhoA and actin, the precise mechanism as well as the underlying actin structures and assembly factors driving the process are unknown. Furthermore, whether specific cell surface receptors trigger entotic invasion in a signal-dependent fashion has not been investigated. In this study, we identify the G-protein-coupled LPA receptor 2 (LPAR2) as a signal transducer specifically required for the actively invading cell during entosis. We find that 12/13G and PDZ-RhoGEF are required for entotic invasion, which is driven by blebbing and a uropod-like actin structure at the rear of the invading cell. Finally, we provide evidence for an involvement of the RhoA-regulated formin Dia1 for entosis downstream of LPAR2. Thus, we delineate a signaling process that regulates actin dynamics during cell-in-cell invasion. DOI: 10.7554/eLife.02786.001 Introduction Entosis has been described as a specialized form of homotypic cell-in-cell invasion in which one cell actively crawls into another (Overholtzer et al., 2007). Frequently, this occurs between tumor cells such as breast, cervical, or colon carcinoma cells and can be triggered by matrix detachment (Overholtzer et al., 2007), suggesting that loss of integrin-mediated adhesion may promote cell-in-cell invasion. -

Identification of Novel Potential Genes Involved in Cancer by Integrated
International Journal of Molecular Sciences Article Identification of Novel Potential Genes Involved in Cancer by Integrated Comparative Analyses Francesco Monticolo 1, Emanuela Palomba 2 and Maria Luisa Chiusano 1,2,* 1 Department of Agricultural Sciences, Università Degli Studi di Napoli Federico II, 80055 Naples, Italy; [email protected] 2 Department of RIMAR, Stazione Zoologica “Anton Dohrn”, 80122 Naples, Italy; [email protected] * Correspondence: [email protected] Received: 26 October 2020; Accepted: 11 December 2020; Published: 15 December 2020 Abstract: The main hallmarks of cancer diseases are the evasion of programmed cell death, uncontrolled cell division, and the ability to invade adjacent tissues. The explosion of omics technologies offers challenging opportunities to identify molecular agents and processes that may play relevant roles in cancer. They can support comparative investigations, in one or multiple experiments, exploiting evidence from one or multiple species. Here, we analyzed gene expression data from induction of programmed cell death and stress response in Homo sapiens and compared the results with Saccharomyces cerevisiae gene expression during the response to cell death. The aim was to identify conserved candidate genes associated with Homo sapiens cell death, favored by crosslinks based on orthology relationships between the two species. Weidentified differentially-expressed genes, pathways that are significantly dysregulated across treatments, and characterized genes among those involved in induced cell death. We investigated on co-expression patterns and identified novel genes that were not expected to be associated with death pathways, that have a conserved pattern of expression between the two species. Finally, we analyzed the resulting list by HumanNet and identified new genes predicted to be involved in cancer. -

A Rhoa and Rnd3 Cycle Regulates Actin Reassembly During Membrane
A RhoA and Rnd3 cycle regulates actin reassembly PNAS PLUS during membrane blebbing Kana Aokia, Fumiyo Maedaa,1, Tomoya Nagasakob,1, Yuki Mochizukia, Seiichi Uchidab, and Junichi Ikenouchia,c,d,2 aDepartment of Biology, Faculty of Sciences, Kyushu University, Fukuoka 819-0395, Japan; bDepartment of Advanced Information Technology, Kyushu University, Fukuoka 819-0395, Japan; cPrecursory Research for Embryonic Science and Technology, Japan Science and Technology Agency, Saitama 332-0012, Japan; and dAMED-PRIME, Japan Agency for Medical Research and Development, Tokyo 100-0004, Japan Edited by Thomas D. Pollard, Yale University, New Haven, CT, and approved February 12, 2016 (received for review January 21, 2016) The actin cytoskeleton usually lies beneath the plasma membrane. membrane blebs is promoted by epidermal growth factor receptor When the membrane-associated actin cytoskeleton is transiently kinase substrate 8 (Eps8) and ezrin, and regulated by a RhoA– disrupted or the intracellular pressure is increased, the plasma Rho-associated protein kinase (ROCK)–Rnd3 feedback loop. membrane detaches from the cortex and protrudes. Such protruded membrane regions are called blebs. However, the molecular mech- Results and Discussion anisms underlying membrane blebbing are poorly understood. This Membrane Blebs Retract from Multiple Sites. We used the human study revealed that epidermal growth factor receptor kinase sub- colon carcinoma cell line DLD1 to observe membrane blebbing. strate 8 (Eps8) and ezrin are important regulators of rapid actin When cultured in 2D conditions, DLD1 cells did not exhibit reassembly for the initiation and retraction of protruded blebs. Live- membrane blebbing (Fig. 1A, Left). However, DLD1 cells ac- cell imaging of membrane blebbing revealed that local reassembly tively formed membrane blebs when embedded in a type I col- of actin filaments occurred at Eps8- and activated ezrin-positive foci lagen gel (Fig. -

Vaccinia Virus Strains Use Distinct Forms of Macropinocytosis for Host-Cell Entry
Vaccinia virus strains use distinct forms of macropinocytosis for host-cell entry Jason Mercer1, Stephan Knébel, Florian I. Schmidt, Josh Crouse, Christine Burkard, and Ari Helenius1 Eidgenössische Technische Hochschule Zurich, Institute of Biochemistry, Eidgenössische Technische Hochschule Hoenggerberg, 8093 Zurich, Switzerland Contributed by Ari Helenius, April 10, 2010 (sent for review January 20, 2010) To enter host cells, vaccinia virus, a prototype poxvirus, can induce Previous studies have suggested that entry of IHD-J MVs might transient macropinocytosis followed by endocytic internalization be different. Instead of blebs, they seem to induce narrow plasma and penetration through the limiting membrane of pinosomes by membrane protrusions (9). The IHD-J virus is also more de- membrane fusion. Although mature virions (MVs) of the Western pendent on glycosaminoglycan binding and does not require reserve (WR) strain do this in HeLa cells by activating transient acidic vacuolar pH (10). To determine whether the two strains of plasma membrane blebbing, MVs from the International Health VACV do, in fact, use different entry strategies, we compared Department-J strain were found to induce rapid formation (and their interaction with HeLa cells and found that the immediate lengthening) of filopodia. When the signaling pathways underly- cellular responses to the two strains were quite different. Clearly ing these responses were compared, differences were observed at the same host cells were capable of activating distinct forms the level of Rho GTPases. Key to the filopodial formation was the of macropinocytosis. virus-induced activation of Cdc42, and for the blebbing response the activation of Rac1. In addition, unlike WR, International Health Results Department-J MVs did not rely on genistein-sensitive tyrosine IHD-J MVs Induce Filopodia. -
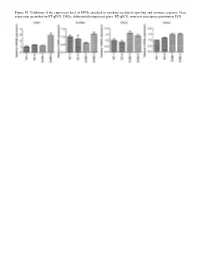
Figure S1. Validation of the Expression Level of Degs Enriched in Cytokine‑Mediated Signaling and Immune Response
Figure S1. Validation of the expression level of DEGs enriched in cytokine‑mediated signaling and immune response. Gene expression quantified by RT‑qPCR. DEGs, differentially expressed genes; RT‑qPCR, reverse‑transcription quantitative PCR. Figure S2. Validation of STAU1‑regulated AS events. IGV‑Sashimi plot revealed (A‑C) three A5SS AS events in three different genes. Reads distribution of each AS event was plotted in the left panel with the transcripts of each gene shown below. The sche‑ matic diagrams depict the structures of two AS events, AS1 (purple line) and AS2 (green line). The exon sequences are denoted by black boxes, the intron sequences by a horizontal line (right panel). RNA‑seq quantification and RT‑qPCR validation of ASEs are presented in the panels on the right. STAU1, double‑stranded RNA‑binding protein Staufen homolog 1; AS, alternative splicing; A5SS, alternative 5'splice site; RNA‑seq, RNA sequencing; RT‑qPCR, reverse‑transcription quantitative PCR. Error bars represent mean ± SEM. *P<0.05. Table SI. Primers used in gene validation experiments. IFIT2‑F CAGCCTACGGCAACTAAA IFIT2‑R GAGCCTTCTCAAAGCACA IFIT3‑F ACACCAAACAATGGCTAC IFIT3‑R TGGACAAACCCTCTAAAC OASL‑F AATGGTGACCGTGATGGG OASL‑R ACCTGAGGATGGAGCAGAG IFI27‑F TTCACTGCGGCGGGAATC IFI27‑R TGGCTGCTATGGAGGACGAG S1PR4‑F TGCTGAAGACGGTGCTGATG S1PR4‑R TGCGGAAGGAGTAGATGATGG CCL5‑F ACGACTGCTGGGTTGGAG CCL5‑R ACCCTGCTGCTTTGCCTA CCL2‑F CTAACCCAGAAACATCCAAT CCL2‑R GCTATGAGCAGCAGGCAC CD44‑F TGGAGGACAGAAAGCCAAGT CD44‑R TTCGCAATGAAACAATCAGTAG PLEKHG2‑M/As‑F CCAAAAGTAAGCCTGTCC PLEKHG2‑M‑R -

Classification of Cell Death
Cell Death and Differentiation (2009) 16, 3–11 & 2009 Macmillan Publishers Limited All rights reserved 1350-9047/09 $32.00 www.nature.com/cdd Review Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009 G Kroemer*,1,2,3, L Galluzzi1,2,3, P Vandenabeele4,5, J Abrams6, ES Alnemri7, EH Baehrecke8, MV Blagosklonny9, WS El-Deiry10, P Golstein11,12,13, DR Green14, M Hengartner15, RA Knight16, S Kumar17, SA Lipton18,19,20, W Malorni21, G Nun˜ez22, ME Peter23, J Tschopp24, J Yuan25, M Piacentini26,27, B Zhivotovsky28 and G Melino29,30 Different types of cell death are often defined by morphological criteria, without a clear reference to precise biochemical mechanisms. The Nomenclature Committee on Cell Death (NCCD) proposes unified criteria for the definition of cell death and of its different morphologies, while formulating several caveats against the misuse of words and concepts that slow down progress in the area of cell death research. Authors, reviewers and editors of scientific periodicals are invited to abandon expressions like ‘percentage apoptosis’ and to replace them with more accurate descriptions of the biochemical and cellular parameters that are actually measured. Moreover, at the present stage, it should be accepted that caspase-independent mechanisms can cooperate with (or substitute for) caspases in the execution of lethal signaling pathways and that ‘autophagic cell death’ is a type of cell death occurring together with (but not necessarily by) autophagic vacuolization. This study details the 2009 recommendations of the NCCD on the use of cell death-related terminology including ‘entosis’, ‘mitotic catastrophe’, ‘necrosis’, ‘necroptosis’ and ‘pyroptosis’. -

Download (PDF)
Table S1. Putative miR-322 target transcripts which is highly expressed in MGCs ________________________________________________________________________________ 0610037L13Rik, 1700037H04Rik, 2310061I04Rik, 2810006K23Rik, Abcc5, Abhd16a, Acbd3, Acox1, Acsbg1, Acsl4, Actr1a, Actr2, Adck5, Adh5, Adrbk1, Aff4, Agk, Ahcyl1, Akap11, Akap7, Akirin1, Alg3, Amfr, Ammecr1, Amotl2, Ankfy1, Ankhd1, Ankrd52, Ap2a1, Ap2b1, Ap3b1, Ap3d1, App, Arcn1, Arf3, Arfgap2, Arhgap12, Arhgap5, Arhgdia, Arhgef11, Arih1, Arl2, Arl3, Arl8b, Armcx6, Asap1, Asnsd1, Atf6, Atg13, Atg4b, Atp13a3, Atp5g1, Atp6v1a, Atxn2, Atxn7l3, Atxn7l3b, AW549877, B4galt1, B4galt7, Bace1, Bag5, Baiap2, Baz2a, BC037034, Bcl2l1, Bcl2l2, Bfar, Bmpr1a, Bptf, Brd2, Brd4, Brpf3, Btbd3, Btg2, Cab39, Cacna2d1, Calm1, Capns1, Caprin1, Capza2, Carm1, Caskin1, Cbfa2t3, Cbx5, Cbx6, Cc2d1b, Ccdc127, Ccdc6, Ccnd2, Ccnt2, Ccnyl1, Cd164, Cd2ap, Cdc25a, Cdc27, Cdc37l1, Cdc42se2, Cdca4, Cdipt, Cdk5rap3, Cdk8, Cdk9, Cdv3, Celf1, Cfl2, Chchd3, Chd6, Chmp1a, Chmp7, Chordc1, Chpf, Chpt1, Chst8, Chtf8, Cisd2, Clasrp, Clcn3, Clstn1, Cmpk1, Cnih2, Cnot1, Cnot2, Col4a3bp, Cope, Cops2, Cops7a, Cops7b, Copz1, Coq6, Cpd, Crebzf, Crim1, Crk, Crkl, Csde1, Cse1l, Ctnnb1, Cul4a, Cul4b, Cxx1a, Cxx1b, Cxx1c, D15Ertd621e, D2hgdh, Dcaf7, Dcbld2, Dcp1a, Dctn5, Ddost, Ddr1, Ddx39, Ddx3x, Ddx6, Dedd, Dhdds, Dhx16, Diap1, Dido1, Dlst, Dmtf1, Dnaja2, Dnajb14, Dnajb2, Dnajc1, Dnajc16, Dnajc25, Dph3, Dpm1, Dpp9, Dpy19l4, Dsel, Dtl, Dvl1, Dync1li2, Dynll2, Dynlt3, Dyrk1a, Dyrk1b, Ebna1bp2, Edc4, Eftud2, Egln2, Eif1a, Eif2b2, Eif2s1,