(F304S) Substitution in the Human ALG6 Gene Is a Common Polymorphism and Not a Causal Mutation of CDG-Ic
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Yeast Genome Gazetteer P35-65
gazetteer Metabolism 35 tRNA modification mitochondrial transport amino-acid metabolism other tRNA-transcription activities vesicular transport (Golgi network, etc.) nitrogen and sulphur metabolism mRNA synthesis peroxisomal transport nucleotide metabolism mRNA processing (splicing) vacuolar transport phosphate metabolism mRNA processing (5’-end, 3’-end processing extracellular transport carbohydrate metabolism and mRNA degradation) cellular import lipid, fatty-acid and sterol metabolism other mRNA-transcription activities other intracellular-transport activities biosynthesis of vitamins, cofactors and RNA transport prosthetic groups other transcription activities Cellular organization and biogenesis 54 ionic homeostasis organization and biogenesis of cell wall and Protein synthesis 48 plasma membrane Energy 40 ribosomal proteins organization and biogenesis of glycolysis translation (initiation,elongation and cytoskeleton gluconeogenesis termination) organization and biogenesis of endoplasmic pentose-phosphate pathway translational control reticulum and Golgi tricarboxylic-acid pathway tRNA synthetases organization and biogenesis of chromosome respiration other protein-synthesis activities structure fermentation mitochondrial organization and biogenesis metabolism of energy reserves (glycogen Protein destination 49 peroxisomal organization and biogenesis and trehalose) protein folding and stabilization endosomal organization and biogenesis other energy-generation activities protein targeting, sorting and translocation vacuolar and lysosomal -

Congenital Disorders of Glycosylation from a Neurological Perspective
brain sciences Review Congenital Disorders of Glycosylation from a Neurological Perspective Justyna Paprocka 1,* , Aleksandra Jezela-Stanek 2 , Anna Tylki-Szyma´nska 3 and Stephanie Grunewald 4 1 Department of Pediatric Neurology, Faculty of Medical Science in Katowice, Medical University of Silesia, 40-752 Katowice, Poland 2 Department of Genetics and Clinical Immunology, National Institute of Tuberculosis and Lung Diseases, 01-138 Warsaw, Poland; [email protected] 3 Department of Pediatrics, Nutrition and Metabolic Diseases, The Children’s Memorial Health Institute, W 04-730 Warsaw, Poland; [email protected] 4 NIHR Biomedical Research Center (BRC), Metabolic Unit, Great Ormond Street Hospital and Institute of Child Health, University College London, London SE1 9RT, UK; [email protected] * Correspondence: [email protected]; Tel.: +48-606-415-888 Abstract: Most plasma proteins, cell membrane proteins and other proteins are glycoproteins with sugar chains attached to the polypeptide-glycans. Glycosylation is the main element of the post- translational transformation of most human proteins. Since glycosylation processes are necessary for many different biological processes, patients present a diverse spectrum of phenotypes and severity of symptoms. The most frequently observed neurological symptoms in congenital disorders of glycosylation (CDG) are: epilepsy, intellectual disability, myopathies, neuropathies and stroke-like episodes. Epilepsy is seen in many CDG subtypes and particularly present in the case of mutations -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Molecular Diagnostic Requisition
BAYLOR MIRACA GENETICS LABORATORIES SHIP TO: Baylor Miraca Genetics Laboratories 2450 Holcombe, Grand Blvd. -Receiving Dock PHONE: 800-411-GENE | FAX: 713-798-2787 | www.bmgl.com Houston, TX 77021-2024 Phone: 713-798-6555 MOLECULAR DIAGNOSTIC REQUISITION PATIENT INFORMATION SAMPLE INFORMATION NAME: DATE OF COLLECTION: / / LAST NAME FIRST NAME MI MM DD YY HOSPITAL#: ACCESSION#: DATE OF BIRTH: / / GENDER (Please select one): FEMALE MALE MM DD YY SAMPLE TYPE (Please select one): ETHNIC BACKGROUND (Select all that apply): UNKNOWN BLOOD AFRICAN AMERICAN CORD BLOOD ASIAN SKELETAL MUSCLE ASHKENAZIC JEWISH MUSCLE EUROPEAN CAUCASIAN -OR- DNA (Specify Source): HISPANIC NATIVE AMERICAN INDIAN PLACE PATIENT STICKER HERE OTHER JEWISH OTHER (Specify): OTHER (Please specify): REPORTING INFORMATION ADDITIONAL PROFESSIONAL REPORT RECIPIENTS PHYSICIAN: NAME: INSTITUTION: PHONE: FAX: PHONE: FAX: NAME: EMAIL (INTERNATIONAL CLIENT REQUIREMENT): PHONE: FAX: INDICATION FOR STUDY SYMPTOMATIC (Summarize below.): *FAMILIAL MUTATION/VARIANT ANALYSIS: COMPLETE ALL FIELDS BELOW AND ATTACH THE PROBAND'S REPORT. GENE NAME: ASYMPTOMATIC/POSITIVE FAMILY HISTORY: (ATTACH FAMILY HISTORY) MUTATION/UNCLASSIFIED VARIANT: RELATIONSHIP TO PROBAND: THIS INDIVIDUAL IS CURRENTLY: SYMPTOMATIC ASYMPTOMATIC *If family mutation is known, complete the FAMILIAL MUTATION/ VARIANT ANALYSIS section. NAME OF PROBAND: ASYMPTOMATIC/POPULATION SCREENING RELATIONSHIP TO PROBAND: OTHER (Specify clinical findings below): BMGL LAB#: A COPY OF ORIGINAL RESULTS ATTACHED IF PROBAND TESTING WAS PERFORMED AT ANOTHER LAB, CALL TO DISCUSS PRIOR TO SENDING SAMPLE. A POSITIVE CONTROL MAY BE REQUIRED IN SOME CASES. REQUIRED: NEW YORK STATE PHYSICIAN SIGNATURE OF CONSENT I certify that the patient specified above and/or their legal guardian has been informed of the benefits, risks, and limitations of the laboratory test(s) requested. -

ALG6-CDG in South Africa: Genotype-Phenotype Description of Five Novel Patients
JIMD Reports DOI 10.1007/8904_2012_150 CASE REPORT ALG6-CDG in South Africa: Genotype-Phenotype Description of Five Novel Patients M. Dercksen • A. C. Crutchley • E. M. Honey • M. M. Lippert • G. Matthijs • L. J. Mienie • H. C. Schuman • B. C. Vorster • J. Jaeken Received: 11 October 2011 /Revised: 30 April 2012 /Accepted: 07 May 2012 # SSIEM and Springer-Verlag Berlin Heidelberg 2012 Abstract ALG6-CDG (formerly named CDG-Ic) (pheno- for the known c.998C>T (p.A333V) mutation and the novel type OMIM 603147, genotype OMIM 604566), is caused by c.1338dupA (p.V447SfsX44) mutation. Four more patients, defective endoplasmic reticulum a-1,3-glucosyltransferase presenting with classical neurological involvement were (E.C 2.4.1.267) in the N-glycan assembly pathway identified and were compound heterozygous for the known (Grunewald€ et al. 2000). It is the second most frequent c.257 + 5G>A splice mutation and the c.680G>A(p.G227E) N-glycosylation disorder after PMM2-CDG; some 37 patients missense mutation. The patients belong to a semi-isolated have been reported with 21 different ALG6 gene mutations Caucasian community that may have originated from Euro- (Haeuptle & Hennet 2009; Al-Owain 2010). We report on the pean pioneers who colonized South Africa in the seventeenth/ clinical and biochemical findings of five novel Caucasian eighteenth centuries. South African patients. The first patient had a severe neuro- gastrointestinal presentation. He was compound heterozygous Introduction Communicated by: Eva Morava, MD PhD ALG6-CG (phenotype OMIM 603147, genotype OMIM Competing interests: none declared : : 604566) is a genetic disorder in the assembly of N-glycans M. -

Viruses Like Sugars: How to Assess Glycan Involvement in Viral Attachment
microorganisms Review Viruses Like Sugars: How to Assess Glycan Involvement in Viral Attachment Gregory Mathez and Valeria Cagno * Institute of Microbiology, Lausanne University Hospital, University of Lausanne, 1011 Lausanne, Switzerland; [email protected] * Correspondence: [email protected] Abstract: The first step of viral infection requires interaction with the host cell. Before finding the specific receptor that triggers entry, the majority of viruses interact with the glycocalyx. Identifying the carbohydrates that are specifically recognized by different viruses is important both for assessing the cellular tropism and for identifying new antiviral targets. Advances in the tools available for studying glycan–protein interactions have made it possible to identify them more rapidly; however, it is important to recognize the limitations of these methods in order to draw relevant conclusions. Here, we review different techniques: genetic screening, glycan arrays, enzymatic and pharmacological approaches, and surface plasmon resonance. We then detail the glycan interactions of enterovirus D68 and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), highlighting the aspects that need further clarification. Keywords: attachment receptor; viruses; glycan; sialic acid; heparan sulfate; HBGA; SARS-CoV-2; EV-D68 Citation: Mathez, G.; Cagno, V. Viruses Like Sugars: How to Assess 1. Introduction Glycan Involvement in Viral This review focuses on methods for assessing the involvement of carbohydrates in Attachment. Microorganisms 2021, 9, viral attachment and entry into the host cell. Viruses often bind to entry receptors that are 1238. https://doi.org/10.3390/ not abundant on the cell surface; to increase their chances of finding them, they initially microorganisms9061238 bind to attachment receptors comprising carbohydrates that are more widely expressed. -

Prenatal Testing Requisition Form
BAYLOR MIRACA GENETICS LABORATORIES SHIP TO: Baylor Miraca Genetics Laboratories 2450 Holcombe, Grand Blvd. -Receiving Dock PHONE: 800-411-GENE | FAX: 713-798-2787 | www.bmgl.com Houston, TX 77021-2024 Phone: 713-798-6555 PRENATAL COMPREHENSIVE REQUISITION FORM PATIENT INFORMATION NAME (LAST,FIRST, MI): DATE OF BIRTH (MM/DD/YY): HOSPITAL#: ACCESSION#: REPORTING INFORMATION ADDITIONAL PROFESSIONAL REPORT RECIPIENTS PHYSICIAN: NAME: INSTITUTION: PHONE: FAX: PHONE: FAX: NAME: EMAIL (INTERNATIONAL CLIENT REQUIREMENT): PHONE: FAX: SAMPLE INFORMATION CLINICAL INDICATION FETAL SPECIMEN TYPE Pregnancy at risk for specific genetic disorder DATE OF COLLECTION: (Complete FAMILIAL MUTATION information below) Amniotic Fluid: cc AMA PERFORMING PHYSICIAN: CVS: mg TA TC Abnormal Maternal Screen: Fetal Blood: cc GESTATIONAL AGE (GA) Calculation for AF-AFP* NTD TRI 21 TRI 18 Other: SELECT ONLY ONE: Abnormal NIPT (attach report): POC/Fetal Tissue, Type: TRI 21 TRI 13 TRI 18 Other: Cultured Amniocytes U/S DATE (MM/DD/YY): Abnormal U/S (SPECIFY): Cultured CVS GA ON U/S DATE: WKS DAYS PARENTAL BLOODS - REQUIRED FOR CMA -OR- Maternal Blood Date of Collection: Multiple Pregnancy Losses LMP DATE (MM/DD/YY): Parental Concern Paternal Blood Date of Collection: Other Indication (DETAIL AND ATTACH REPORT): *Important: U/S dating will be used if no selection is made. Name: Note: Results will differ depending on method checked. Last Name First Name U/S dating increases overall screening performance. Date of Birth: KNOWN FAMILIAL MUTATION/DISORDER SPECIFIC PRENATAL TESTING Notice: Prior to ordering testing for any of the disorders listed, you must call the lab and discuss the clinical history and sample requirements with a genetic counselor. -

Genetic Predisposition to Fetal Alcohol Syndrome: Association with Congenital Disorders of N-Glycosylation
nature publishing group Basic Science Investigation | Articles Genetic predisposition to fetal alcohol syndrome: association with congenital disorders of N-glycosylation María E. de la Morena-Barrio1, María J. Ballesta-Martínez2, Raquel López-Gálvez1, Ana I. Antón1, Vanessa López-González2, Laia Martínez-Ribot3,JoséPadilla1, Antonia Miñano1, Oscar García-Algar4, Miguel Del Campo5, Javier Corral1, Encarna Guillén-Navarro2 and Vicente Vicente1 BACKGROUND: Fetal alcohol syndrome (FAS) is caused by exposure, with the most serious consequences in those fetuses maternal alcohol consumption during pregnancy; although exposed to high concentrations during the initial weeks of additional factors might be involved, as development and pregnancy (6–9); however, additional factors may be involved severity are not directly related to alcohol intake. The as some exposed pregnancies develop FAS, whereas others do abnormal glycosylation caused by alcohol might play a role not, and the severity of patients is highly heterogeneous. The in FAS according to the clinical similarities shared with estimated prevalence of FAS is 1–3/1,000 live births and congenital disorders of glycosylation (CDG). Thus, mutations constitutes the principal cause of non-inherited mental underlying CDG, affecting genes involved in glycosylation, disability (6,7,10,11). could also be involved in FAS. Diverse pathophysiological mechanisms have been pro- METHODS: A panel of 74 genes involved in N-glycosylation posed to underlie FAS, but none of them completely clarifies was sequenced in 25 FAS patients and 20 controls with this disorder. Recently, a new pathogenetic model suggests prenatal alcohol exposure. Transferrin glycoforms were that glycosylation defects underlie FAS based on the evaluated by HPLC. -

F04b57ca351b0dc3389ebe992c
Review Insights into complexity of congenital disorders of glycosylation Sandra Supraha Goreta*, Sanja Dabelic, Jerka Dumic University of Zagreb, Faculty of Pharmacy and Biochemistry, Department of Biochemistry and Molecular Biology, Zagreb, Croatia *Corresponding author: [email protected] Abstract Biochemical and biological properties of glycoconjugates are strongly determined by the specifi c structure of its glycan parts. Glycosylation, the covalent attachment of sugars to proteins and lipids, is very complex and highly-coordinated process involving > 250 gene products. Defi ciency of glycosylation enzymes or transporters results in impaired glycosylation, and consequently pathological modulation of many physiological processes. Inborn defects of glycosylation enzymes, caused by the specifi cmutations, lead to the development of rare, but severe diseases – congenital disor- ders of glycosylation (CDGs). Up today, there are more than 45 known CDGs. Their clinical manifestations range from very mild to extremely severe (even lethal) and unfortunately, only three of them can be eff ectively treated nowadays. CDG symptoms highly vary, though someare common for several CDG types but also for other unrelated diseases, especially neurological ones, leaving the possibility that many CDGs cases are under- or mis- diagnosed. Glycan analysis of serum transferrin (by isoelectric focusing or more sophisticated methods, such as HPLC (high-performance liquid chro- matography) or MALDI (matrix-assisted laser desorption/ionization)) or serum N-glycans -

Mutations in PHKA1, PHKG1 Or Six Other Candidate Genes Explain Only a Minority of Cases
European Journal of Human Genetics (2003) 11, 516–526 & 2003 Nature Publishing Group All rights reserved 1018-4813/03 $25.00 www.nature.com/ejhg ARTICLE Muscle glycogenosis with low phosphorylase kinase activity: mutations in PHKA1, PHKG1 or six other candidate genes explain only a minority of cases Barbara Burwinkel1,7, Bin Hu1, Anja Schroers2, Paula R. Clemens3,8, Shimon W. Moses4, Yoon S. Shin5, Dieter Pongratz6, Matthias Vorgerd2 and Manfred W. Kilimann*,1,9 1Institut fu¨r Physiologische Chemie, Ruhr-Universita¨t Bochum, D-44780 Bochum, Germany; 2Neurologische Universita¨tsklinik Bergmannsheil, Ruhr-Universita¨t Bochum, Bu¨rkle-de-la-Camp-Platz 1, D-44789 Bochum, Germany; 3Department of Neurology, Mayo Clinic, Rochester, MN 55905, USA; 4Department of Pediatrics, Soroka Medical Center, Ben Gurion University of the Negev, IL-84105 Beer-Sheva, Israel; 5Stoffwechselzentrum, Dr V Haunersches Kinderspital der Universita¨t Mu¨nchen, D-80337 Mu¨nchen, Germany; 6Friedrich-Baur-Institut der Universita¨t Mu¨nchen, Ziemssenstr. 1, D-80336 Mu¨nchen, Germany Muscle-specific deficiency of phosphorylase kinase (Phk) causes glycogen storage disease, clinically manifesting in exercise intolerance with early fatiguability, pain, cramps and occasionally myoglobinuria. In two patients and in a mouse mutant with muscle Phk deficiency, mutations were previously found in the muscle isoform of the Phk a subunit, encoded by the X-chromosomal PHKA1 gene (MIM # 311870). No mutations have been identified in the muscle isoform of the Phk c subunit (PHKG1). In the present study, we determined the structure of the PHKG1 gene and characterized its relationship to several pseudogenes. In six patients with adult- or juvenile-onset muscle glycogenosis and low Phk activity, we then searched for mutations in eight candidate genes. -

Genome-Wide Investigation of Cellular Functions for Trna Nucleus
Genome-wide Investigation of Cellular Functions for tRNA Nucleus- Cytoplasm Trafficking in the Yeast Saccharomyces cerevisiae DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Hui-Yi Chu Graduate Program in Molecular, Cellular and Developmental Biology The Ohio State University 2012 Dissertation Committee: Anita K. Hopper, Advisor Stephen Osmani Kurt Fredrick Jane Jackman Copyright by Hui-Yi Chu 2012 Abstract In eukaryotic cells tRNAs are transcribed in the nucleus and exported to the cytoplasm for their essential role in protein synthesis. This export event was thought to be unidirectional. Surprisingly, several lines of evidence showed that mature cytoplasmic tRNAs shuttle between nucleus and cytoplasm and their distribution is nutrient-dependent. This newly discovered tRNA retrograde process is conserved from yeast to vertebrates. Although how exactly the tRNA nuclear-cytoplasmic trafficking is regulated is still under investigation, previous studies identified several transporters involved in tRNA subcellular dynamics. At least three members of the β-importin family function in tRNA nuclear-cytoplasmic intracellular movement: (1) Los1 functions in both the tRNA primary export and re-export processes; (2) Mtr10, directly or indirectly, is responsible for the constitutive retrograde import of cytoplasmic tRNA to the nucleus; (3) Msn5 functions solely in the re-export process. In this thesis I focus on the physiological role(s) of the tRNA nuclear retrograde pathway. One possibility is that nuclear accumulation of cytoplasmic tRNA serves to modulate translation of particular transcripts. To test this hypothesis, I compared expression profiles from non-translating mRNAs and polyribosome-bound translating mRNAs collected from msn5Δ and mtr10Δ mutants and wild-type cells, in fed or acute amino acid starvation conditions. -
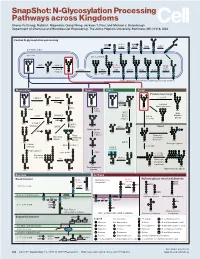
N-Glycosylation Processing Pathways Across Kingdoms Cheng-Yu Chung, Natalia I
SnapShot: N-Glycosylation Processing Pathways across Kingdoms Cheng-Yu Chung, Natalia I. Majewska, Qiong Wang, Jackson T. Paul, and Michael J. Betenbaugh Department of Chemical and Biomolecular Engineering, The Johns Hopkins University, Baltimore, MD 21218, USA Central N-glycosylation processing UDP ALG13 GDP ALG7 ALG14 ALG1 ALG2 ALG11 P P P P P CYTOPLASM P P P P P P GOLGI ER LUMEN GNTI α -Man IA En Bloc ALG3 Transfer ALG9 α-Man IB ER Man I α -Glc I OST ALG10 ALG8 ALG6 ALG12 Asn α -Man IC Asn Asn Asn α -Glc II P P P P P Asn P P P P P ALGs: asparagine-linked N-glycosylation processing enzymes Mammalian Insect Plant Fungi Filamentous fungi α-Man II FUT8 α-1,2-MT XYLT GDP Terminal Asn Asn Asn Asn UDP Asn Galactofuranose GNTII Asn Asn FUT8 f ± α-Gal Asn FUTC3 GNTII High FUT11 α -GALT B14GALTI Och1p Mannose FUT12 glycan α-Man II α-1,2-MT UDP Asn Asn ST3GAL4 Asn Asn GNTIV ST6GAL1 GNTIII GNTV Asn GNase Yeast Hybrid glycan P CMP GNase Asn Asn Tetra- α-Man II Mannosyl- Bisecting antennary phosphate glycan glycan transferase Asn Asn Asn Asn GALT1 Asn ST8SIA2 Core type glycan GNTs B14GALTI α-1,6-MT ST8SIA4 Asn Lewis A structure Polysialylation α-Man PolyLacNAc α-1,2-MT Alternative α-1,3-MT P sialic acid FUT13 ] [ STs Mannosyl- n phosphate Asn Asn Asn transferase Asn Asn Paucimannose Hypermannose glycan Asn Asn Asn glycans Bacteria Archaea Archaea glycan structural diversity Block transfer En Bloc Methanococcus En Bloc Transfer Transfer maripaludis OCH3 Thr OCH AgIB Thr OCH3 3 PERIPLASM PglB B B Asn P - P Asn OSO3 P Asn Asn INNER MEMBRANE P Asn P