Janssen Ad26.COV2.S Vaccine for the Prevention of COVID-19
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Vaccination Certificate Information Version: 21 July 2021
معلومات عن شهادة التطعيم، بالعربية 中文疫苗接种凭证信息 Informations sur le certificat de vaccination en FRANÇAIS Информация о сертификате вакцинации на РУССКОМ ЯЗЫКЕ Información sobre el Certificado de Vacunación en ESPAÑOL UN SYSTEM-WIDE COVID-19 VACCINATION PROGRAMME VACCINATION CERTIFICATE INFORMATION VERSION: 21 JULY 2021 ABOUT THE UN SYSTEM-WIDE COVID-19 VACCINATION PROGRAMME The UN is committed to ensuring the protection of its personnel. Tasked by the Secretary-General, the Department of Operational Support (DOS) is leading a coordinated, UN-system-wide effort to ensure the availability of vaccine to UN personnel, their dependents and implementing partners. The roll-out of the UN System-wide COVID-19 Vaccination Programme (the “Programme”) for UN personnel will provide a significant boost to the ability of personnel to stay and deliver on the Organization's mandates, to support beneficiaries in the communities they serve, and to contribute to our on-going work to recover better together from the pandemic. Information about the Programme is available at: https://www.un.org/en/coronavirus/vaccination ABOUT THE VACCINATION CERTIFICATE Upon administration of the requisite vaccine dosage, a certificate of vaccination is generated through the Programme’s registration platform (the “Platform”). The certificate generated by the Platform uses a standardized format, similar to the one used by the WHO in its “International Certificate of Vaccination or Prophylaxis” (Yellow) vaccination booklet. Note: This certificate may not dispense with travel restrictions put in place by the country(ies) of destination. As the nature of the pandemic continues to rapidly evolve, it is highly advisable to check the latest travel regulations. -

Global Dynamics of a Vaccination Model for Infectious Diseases with Asymptomatic Carriers
MATHEMATICAL BIOSCIENCES doi:10.3934/mbe.2016019 AND ENGINEERING Volume 13, Number 4, August 2016 pp. 813{840 GLOBAL DYNAMICS OF A VACCINATION MODEL FOR INFECTIOUS DISEASES WITH ASYMPTOMATIC CARRIERS Martin Luther Mann Manyombe1;2 and Joseph Mbang1;2 Department of Mathematics, Faculty of Science University of Yaounde 1, P.O. Box 812 Yaounde, Cameroon 1;2;3; Jean Lubuma and Berge Tsanou ∗ Department of Mathematics and Applied Mathematics University of Pretoria, Pretoria 0002, South Africa (Communicated by Abba Gumel) Abstract. In this paper, an epidemic model is investigated for infectious dis- eases that can be transmitted through both the infectious individuals and the asymptomatic carriers (i.e., infected individuals who are contagious but do not show any disease symptoms). We propose a dose-structured vaccination model with multiple transmission pathways. Based on the range of the explic- itly computed basic reproduction number, we prove the global stability of the disease-free when this threshold number is less or equal to the unity. Moreover, whenever it is greater than one, the existence of the unique endemic equilibrium is shown and its global stability is established for the case where the changes of displaying the disease symptoms are independent of the vulnerable classes. Further, the model is shown to exhibit a transcritical bifurcation with the unit basic reproduction number being the bifurcation parameter. The impacts of the asymptomatic carriers and the effectiveness of vaccination on the disease transmission are discussed through through the local and the global sensitivity analyses of the basic reproduction number. Finally, a case study of hepatitis B virus disease (HBV) is considered, with the numerical simulations presented to support the analytical results. -

COVID-19 Vaccines: Update on Allergic Reactions, Contraindications, and Precautions
Centers for Disease Control and Prevention Center for Preparedness and Response COVID-19 Vaccines: Update on Allergic Reactions, Contraindications, and Precautions Clinician Outreach and Communication Activity (COCA) Webinar Wednesday, December 30, 2020 Continuing Education Continuing education will not be offered for this COCA Call. To Ask a Question ▪ All participants joining us today are in listen-only mode. ▪ Using the Webinar System – Click the “Q&A” button. – Type your question in the “Q&A” box. – Submit your question. ▪ The video recording of this COCA Call will be posted at https://emergency.cdc.gov/coca/calls/2020/callinfo_123020.asp and available to view on-demand a few hours after the call ends. ▪ If you are a patient, please refer your questions to your healthcare provider. ▪ For media questions, please contact CDC Media Relations at 404-639-3286, or send an email to [email protected]. Centers for Disease Control and Prevention Center for Preparedness and Response Today’s First Presenter Tom Shimabukuro, MD, MPH, MBA CAPT, U.S. Public Health Service Vaccine Safety Team Lead COVID-19 Response Centers for Disease Control and Prevention Centers for Disease Control and Prevention Center for Preparedness and Response Today’s Second Presenter Sarah Mbaeyi, MD, MPH CDR, U.S. Public Health Service Clinical Guidelines Team COVID-19 Response Centers for Disease Control and Prevention National Center for Immunization & Respiratory Diseases Anaphylaxis following mRNA COVID-19 vaccination Tom Shimabukuro, MD, MPH, MBA CDC COVID-19 Vaccine -

Pricing*, Pool and Payment** Due Dates January - December 2021 Mideast Marketing Area Federal Order No
Pricing*, Pool and Payment** Due Dates January - December 2021 Mideast Marketing Area Federal Order No. 33 Class & Market Administrator Payment Dates for Producer Milk Component Final Pool Producer Advance Prices Payment Dates Final Payment Due Partial Payment Due Pool Month Prices Release Date Payrolls Due & Pricing Factors PSF, Admin., MS Cooperative Nonmember Cooperative Nonmember January February 3 * February 13 February 22 December 23, 2020 February 16 ** February 16 February 17 Janaury 25 January 26 February March 3 * March 13 March 22 January 21 * March 15 March 16 March 17 February 25 February 26 March March 31 * April 13 April 22 February 18 * April 15 April 16 April 19 ** March 25 March 26 April May 5 May 13 May 22 March 17 * May 17 ** May 17 ** May 17 April 26 ** April 26 May June 3 * June 13 June 22 April 21 * June 15 June 16 June 17 May 25 May 26 June June 30 * July 13 July 22 May 19 * July 15 July 16 July 19 ** June 25 June 28 ** July August 4 * August 13 August 22 June 23 August 16 ** August 16 August 17 July 26 ** July 26 August September 1 * September 13 September 22 July 21 * September 15 September 16 September 17 August 25 August 26 September September 29 * October 13 October 22 August 18 * October 15 October 18 ** October 18 ** September 27 ** September 27 ** October November 3 * November 13 November 22 September 22 * November 15 November 16 November 17 October 25 October 26 November December 1 * December 13 December 22 October 20 * December 15 December 16 December 17 November 26 ** November 26 December January 5, 2022 January 13, 2022 January 22, 2022 November 17 * January 18, 2022 ** January 18, 2022 ** January 18, 2022 ** December 27 ** December 27 ** * If the release date does not fall on the 5th (Class & Component Prices) or 23rd (Advance Prices & Pricing Factors), the most current release preceding will be used in the price calculation. -

Considerations for Causality Assessment of Neurological And
Occasional essay J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp-2021-326924 on 6 August 2021. Downloaded from Considerations for causality assessment of neurological and neuropsychiatric complications of SARS- CoV-2 vaccines: from cerebral venous sinus thrombosis to functional neurological disorder Matt Butler ,1 Arina Tamborska,2,3 Greta K Wood,2,3 Mark Ellul,4 Rhys H Thomas,5,6 Ian Galea ,7 Sarah Pett,8 Bhagteshwar Singh,3 Tom Solomon,4 Thomas Arthur Pollak,9 Benedict D Michael,2,3 Timothy R Nicholson10 For numbered affiliations see INTRODUCTION More severe potential adverse effects in the open- end of article. The scientific community rapidly responded to label phase of vaccine roll- outs are being collected the COVID-19 pandemic by developing novel through national surveillance systems. In the USA, Correspondence to SARS- CoV-2 vaccines (table 1). As of early June Dr Timothy R Nicholson, King’s roughly 372 adverse events have been reported per College London, London WC2R 2021, an estimated 2 billion doses have been million doses, which is a lower rate than expected 1 2LS, UK; timothy. nicholson@ administered worldwide. Neurological adverse based on the clinical trials.6 kcl. ac. uk events following immunisation (AEFI), such as In the UK, adverse events are reported via the cerebral venous sinus thrombosis and demyelin- MB and AT are joint first Coronavirus Yellow Card reporting website. As of ating episodes, have been reported. In some coun- authors. early June 2021, approximately 250 000 Yellow tries, these have led to the temporary halting of BDM and TRN are joint senior Cards have been submitted, equating to around authors. -
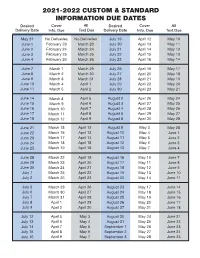
2021-2022 Custom & Standard Information Due Dates
2021-2022 CUSTOM & STANDARD INFORMATION DUE DATES Desired Cover All Desired Cover All Delivery Date Info. Due Text Due Delivery Date Info. Due Text Due May 31 No Deliveries No Deliveries July 19 April 12 May 10 June 1 February 23 March 23 July 20 April 13 May 11 June 2 February 24 March 24 July 21 April 14 May 12 June 3 February 25 March 25 July 22 April 15 May 13 June 4 February 26 March 26 July 23 April 16 May 14 June 7 March 1 March 29 July 26 April 19 May 17 June 8 March 2 March 30 July 27 April 20 May 18 June 9 March 3 March 31 July 28 April 21 May 19 June 10 March 4 April 1 July 29 April 22 May 20 June 11 March 5 April 2 July 30 April 23 May 21 June 14 March 8 April 5 August 2 April 26 May 24 June 15 March 9 April 6 August 3 April 27 May 25 June 16 March 10 April 7 August 4 April 28 May 26 June 17 March 11 April 8 August 5 April 29 May 27 June 18 March 12 April 9 August 6 April 30 May 28 June 21 March 15 April 12 August 9 May 3 May 28 June 22 March 16 April 13 August 10 May 4 June 1 June 23 March 17 April 14 August 11 May 5 June 2 June 24 March 18 April 15 August 12 May 6 June 3 June 25 March 19 April 16 August 13 May 7 June 4 June 28 March 22 April 19 August 16 May 10 June 7 June 29 March 23 April 20 August 17 May 11 June 8 June 30 March 24 April 21 August 18 May 12 June 9 July 1 March 25 April 22 August 19 May 13 June 10 July 2 March 26 April 23 August 20 May 14 June 11 July 5 March 29 April 26 August 23 May 17 June 14 July 6 March 30 April 27 August 24 May 18 June 15 July 7 March 31 April 28 August 25 May 19 June 16 July 8 April 1 April 29 August 26 May 20 June 17 July 9 April 2 April 30 August 27 May 21 June 18 July 12 April 5 May 3 August 30 May 24 June 21 July 13 April 6 May 4 August 31 May 25 June 22 July 14 April 7 May 5 September 1 May 26 June 23 July 15 April 8 May 6 September 2 May 27 June 24 July 16 April 9 May 7 September 3 May 28 June 25. -

Electronic Support for Public Health–Vaccine Adverse Event Reporting System (ESP:VAERS)
Grant Final Report Grant ID: R18 HS 017045 Electronic Support for Public Health–Vaccine Adverse Event Reporting System (ESP:VAERS) Inclusive dates: 12/01/07 - 09/30/10 Principal Investigator: Lazarus, Ross, MBBS, MPH, MMed, GDCompSci Team members: Michael Klompas, MD, MPH Performing Organization: Harvard Pilgrim Health Care, Inc. Project Officer: Steve Bernstein Submitted to: The Agency for Healthcare Research and Quality (AHRQ) U.S. Department of Health and Human Services 540 Gaither Road Rockville, MD 20850 www.ahrq.gov Abstract Purpose: To develop and disseminate HIT evidence and evidence-based tools to improve healthcare decision making through the use of integrated data and knowledge management. Scope: To create a generalizable system to facilitate detection and clinician reporting of vaccine adverse events, in order to improve the safety of national vaccination programs. Methods: Electronic medical records available from all ambulatory care encounters in a large multi-specialty practice were used. Every patient receiving a vaccine was automatically identified, and for the next 30 days, their health care diagnostic codes, laboratory tests, and medication prescriptions were evaluated for values suggestive of an adverse event. Results: Restructuring at CDC and consequent delays in terms of decision making have made it challenging despite best efforts to move forward with discussions regarding the evaluation of ESP:VAERS performance in a randomized trial and comparison of ESP:VAERS performance to existing VAERS and Vaccine Safety Datalink data. However, Preliminary data were collected and analyzed and this initiative has been presented at a number of national symposia. Key Words: electronic health records, vaccinations, adverse event reporting The authors of this report are responsible for its content. -

Vaccine Hesitancy
WHY CHILDREN WORKSHOP ON IMMUNIZATIONS ARE NOT VACCINATED? VACCINE HESITANCY José Esparza MD, PhD - Adjunct Professor, Institute of Human Virology, University of Maryland School of Medicine, Baltimore, MD, USA - Robert Koch Fellow, Robert Koch Institute, Berlin, Germany - Senior Advisor, Global Virus Network, Baltimore, MD, USA. Formerly: - Bill & Melinda Gates Foundation, Seattle, WA, USA - World Health Organization, Geneva, Switzerland The value of vaccination “The impact of vaccination on the health of the world’s people is hard to exaggerate. With the exception of safe water, no other modality has had such a major effect on mortality reduction and population growth” Stanley Plotkin (2013) VACCINES VAILABLE TO PROTECT AGAINST MORE DISEASES (US) BASIC VACCINES RECOMMENDED BY WHO For all: BCG, hepatitis B, polio, DTP, Hib, Pneumococcal (conjugated), rotavirus, measles, rubella, HPV. For certain regions: Japanese encephalitis, yellow fever, tick-borne encephalitis. For some high-risk populations: typhoid, cholera, meningococcal, hepatitis A, rabies. For certain immunization programs: mumps, influenza Vaccines save millions of lives annually, worldwide WHAT THE WORLD HAS ACHIEVED: 40 YEARS OF INCREASING REACH OF BASIC VACCINES “Bill Gates Chart” 17 M GAVI 5.6 M 4.2 M Today (ca 2015): <5% of children in GAVI countries fully immunised with the 11 WHO- recommended vaccines Seth Berkley (GAVI) The goal: 50% of children in GAVI countries fully immunised by 2020 Seth Berkley (GAVI) The current world immunization efforts are achieving: • Equity between high and low-income countries • Bringing the power of vaccines to even the world’s poorest countries • Reducing morbidity and mortality in developing countries • Eliminating and eradicating disease WHY CHILDREN ARE NOT VACCINATED? •Vaccines are not available •Deficient health care systems •Poverty •Vaccine hesitancy (reticencia a la vacunacion) VACCINE HESITANCE: WHO DEFINITION “Vaccine hesitancy refers to delay in acceptance or refusal of vaccines despite availability of vaccination services. -
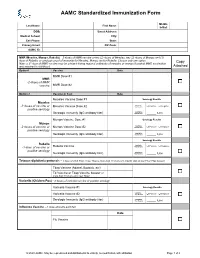
AAMC Standardized Immunization Form
AAMC Standardized Immunization Form Middle Last Name: First Name: Initial: DOB: Street Address: Medical School: City: Cell Phone: State: Primary Email: ZIP Code: AAMC ID: MMR (Measles, Mumps, Rubella) – 2 doses of MMR vaccine or two (2) doses of Measles, two (2) doses of Mumps and (1) dose of Rubella; or serologic proof of immunity for Measles, Mumps and/or Rubella. Choose only one option. Copy Note: a 3rd dose of MMR vaccine may be advised during regional outbreaks of measles or mumps if original MMR vaccination was received in childhood. Attached Option1 Vaccine Date MMR Dose #1 MMR -2 doses of MMR vaccine MMR Dose #2 Option 2 Vaccine or Test Date Measles Vaccine Dose #1 Serology Results Measles Qualitative -2 doses of vaccine or Measles Vaccine Dose #2 Titer Results: Positive Negative positive serology Quantitative Serologic Immunity (IgG antibody titer) Titer Results: _____ IU/ml Mumps Vaccine Dose #1 Serology Results Mumps Qualitative -2 doses of vaccine or Mumps Vaccine Dose #2 Titer Results: Positive Negative positive serology Quantitative Serologic Immunity (IgG antibody titer) Titer Results: _____ IU/ml Serology Results Rubella Qualitative Positive Negative -1 dose of vaccine or Rubella Vaccine Titer Results: positive serology Quantitative Serologic Immunity (IgG antibody titer) Titer Results: _____ IU/ml Tetanus-diphtheria-pertussis – 1 dose of adult Tdap; if last Tdap is more than 10 years old, provide date of last Td or Tdap booster Tdap Vaccine (Adacel, Boostrix, etc) Td Vaccine or Tdap Vaccine booster (if more than 10 years since last Tdap) Varicella (Chicken Pox) - 2 doses of varicella vaccine or positive serology Varicella Vaccine #1 Serology Results Qualitative Varicella Vaccine #2 Titer Results: Positive Negative Serologic Immunity (IgG antibody titer) Quantitative Titer Results: _____ IU/ml Influenza Vaccine --1 dose annually each fall Date Flu Vaccine © 2020 AAMC. -

COVID-19 Vaccineadmin VAERS
Reporting a Suspected Vaccine Adverse Event Indian Health Service, National Pharmacy & Therapeutics Committee CAPT Matthew Clark, MD, FAAP, FACP, Chair CAPT Ryan Schupbach, PharmD, BCPS, CACP, Vice Chair CAPT Christopher Lamer, PharmD, MHS, BCPS, CDE, Director of Pharmacovigilance October 2020 Potential New Vaccine for COVID-19 • Operation Warp Speed (OWS) • Collaboration between the Federal Government and biopharmaceutical companies to develop medications, diagnostic tests, and vaccines. • Shortened timelines but safety and efficacy are the primary focus. • Vaccines must be at least 50% effective for FDA EUA or approval. • Vaccines must be safe, and benefits of immunization must outweigh any risks of adverse events. • Continued need for influenza immunization • Continued need for scheduled immunizations https://www.hhs.gov/coronavirus/explaining-operation-warp-speed/index.html 2 Vaccine Safety: Common Adverse Events Vaccines are considered to be safe and effective with most common adverse events being mild and are signs that the body is developing immunity: • Pain, swelling, or redness where the shot was given • Mild fever • Chills • Feeling tired • Headache • Muscle and joint aches https://www.vaccines.gov/basics/safety/side_effects 3 Vaccine Safety: Serious Adverse Events More serious side effects are rare but can occur. Some examples are: • Anaphylaxis (0.65 cases/1 million vaccinations) • Thrombocytopenia from Rubella vaccine (1 case/40,000 vaccinations) • Orchitis from Mumps vaccine (0.3 cases/1 million vaccinations) • Intussusception from Rotavirus vaccine (1 case/100,000 vaccinations) • Guillain-Barre from flu vaccine (1 case/1.25 million vaccinations; association is stronger with flu infection than the vaccine) Spencer JP, Trondsen Pawlowski RH, Thomas S. Vaccine Adverse Events: Separating Myth from Reality. -

Statements Contained in This Release As the Result of New Information Or Future Events Or Developments
Pfizer and BioNTech Provide Update on Booster Program in Light of the Delta-Variant NEW YORK and MAINZ, GERMANY, July 8, 2021 — As part of Pfizer’s and BioNTech’s continued efforts to stay ahead of the virus causing COVID-19 and circulating mutations, the companies are providing an update on their comprehensive booster strategy. Pfizer and BioNTech have seen encouraging data in the ongoing booster trial of a third dose of the current BNT162b2 vaccine. Initial data from the study demonstrate that a booster dose given 6 months after the second dose has a consistent tolerability profile while eliciting high neutralization titers against the wild type and the Beta variant, which are 5 to 10 times higher than after two primary doses. The companies expect to publish more definitive data soon as well as in a peer-reviewed journal and plan to submit the data to the FDA, EMA and other regulatory authorities in the coming weeks. In addition, data from a recent Nature paper demonstrate that immune sera obtained shortly after dose 2 of the primary two dose series of BNT162b2 have strong neutralization titers against the Delta variant (B.1.617.2 lineage) in laboratory tests. The companies anticipate that a third dose will boost those antibody titers even higher, similar to how the third dose performs for the Beta variant (B.1.351). Pfizer and BioNTech are conducting preclinical and clinical tests to confirm this hypothesis. While Pfizer and BioNTech believe a third dose of BNT162b2 has the potential to preserve the highest levels of protective efficacy against all currently known variants including Delta, the companies are remaining vigilant and are developing an updated version of the Pfizer-BioNTech COVID-19 vaccine that targets the full spike protein of the Delta variant. -

COVID-19 Vaccines Frequently Asked Questions
Page 1 of 12 COVID-19 Vaccines 2020a Frequently Asked Questions Michigan.gov/Coronavirus The information in this document will change frequently as we learn more about COVID-19 vaccines. There is a lot we are learning as the pandemic and COVID-19 vaccines evolve. The approach in Michigan will adapt as we learn more. September 29, 2021. Quick Links What’s new | Why COVID-19 vaccination is important | Booster and additional doses | What to expect when you get vaccinated | Safety of the vaccine | Vaccine distribution/prioritization | Additional vaccine information | Protecting your privacy | Where can I get more information? What’s new − Pfizer booster doses recommended for some people to boost waning immunity six months after completing the Pfizer vaccine. Why COVID-19 vaccination is important − If you are fully vaccinated, you don’t have to quarantine after being exposed to COVID-19, as long as you don’t have symptoms. This means missing less work, school, sports and other activities. − COVID-19 vaccination is the safest way to build protection. COVID-19 is still a threat, especially to people who are unvaccinated. Some people who get COVID-19 can become severely ill, which could result in hospitalization, and some people have ongoing health problems several weeks or even longer after getting infected. Even people who did not have symptoms when they were infected can have these ongoing health problems. − After you are fully vaccinated for COVID-19, you can resume many activities that you did before the pandemic. CDC recommends that fully vaccinated people wear a mask in public indoor settings if they are in an area of substantial or high transmission.