At the Gabaa Receptor
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(12) Patent Application Publication (10) Pub. No.: US 2017/0020892 A1 Thompson Et Al
US 20170020892A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2017/0020892 A1 Thompson et al. (43) Pub. Date: Jan. 26, 2017 (54) USE OF NEGATIVE MODULATORS OF Related U.S. Application Data GABA RECEPTORS CONTAINING ALPHAS SUBUNITS AS FAST ACTING (60) Provisional application No. 61/972,446, filed on Mar. ANTDEPRESSANTS 31, 2014. (71) Applicant: University of Maryland, Baltimore, Publication Classification Baltimore, MD (US) (51) Int. Cl. A 6LX 3/557 (2006.01) (72) Inventors: Scott Thompson, Baltimore, MD (US); A6II 3/53 (2006.01) Mark D. Kvarta, Ellicott City, MD A6II 45/06 (2006.01) (US); Adam Van Dyke, Baltimore, MD (52) U.S. Cl. (US) CPC ........... A61 K3I/55.17 (2013.01); A61K 45/06 (2013.01); A61 K3I/53 (2013.01) (73) Assignee: University of Maryland, Baltimore, Baltimore, MD (US) (57) ABSTRACT Embodiments of the disclosure include methods and com (21) Appl. No.: 15/300,984 positions related to treatment of one or more medical conditions with one or more negative modulators of GABA (22) PCT Filed: Mar. 31, 2015 receptors. In specific embodiments, depression and/or Sui cidability is treated or ameliorated or prevented with one or (86) PCT No.: PCT/US2O15/023667 more negative modulators of GABA receptors, such as a S 371 (c)(1), partial inverse agonist of a GABA receptor comprising an (2) Date: Sep. 30, 2016 alpha5 subunit. Patent Application Publication Jan. 26, 2017. Sheet 1 of 12 US 2017/002O892 A1 ×1/ /|\ Patent Application Publication Jan. 26, 2017. Sheet 3 of 12 US 2017/002O892 A1 & Patent Application Publication Jan. -
ANNNNNNNNNNNNNNNNNNNN 100A 006 Left Eye Input Right Eye Input
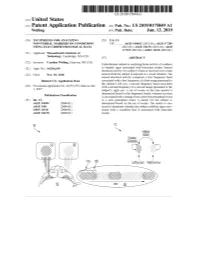
US 20190175049A1 ( 19) United States (12 ) Patent Application Publication (10 ) Pub. No. : US 2019 /0175049 A1 Welling ( 43 ) Pub . Date : Jun . 13 , 2019 ( 54 ) TECHNIQUES FOR ANALYZING (52 ) U . S . CI. NON -VERBAL MARKERS OF CONDITIONS CPC . .. A61B 5 /04842 (2013 . 01 ) ; A61B 5 / 7289 USING ELECTROPHYSIOLOGICAL DATA (2013 . 01) ; A61B 5 /0478 ( 2013 .01 ) ; A61B 5 /7225 ( 2013. 01 ) ; G06N 20 / 10 (2019 .01 ) (71 ) Applicant: Massachusetts Institute of Technology , Cambridge , MA (US ) ( 57 ) ABSTRACT (72 ) Inventor : Caroline Welling, Hanover, NH (US ) Embodiments related to analyzing brain activity of a subject to identify signs associated with binocular rivalry . Sensed ( 21 ) Appl. No. : 16 / 206, 639 electrical activity of a subject' s brain is received over a time period while the subject is exposed to a visual stimulus. The ( 22 ) Filed : Nov. 30 , 2018 sensed electrical activity comprises a first frequency band Related U . S . Application Data associated with a first frequency of a first image presented to the subject ' s left eye , a second frequency band associated (60 ) Provisional application No .62 / 593 , 535, filed on Dec . with a second frequency of a second image presented to the 1 , 2017 subject ' s right eye . A set of events in the time period is determined based on the frequency bands, wherein an event Publication Classification is associated with a change from a previous perceptual event (51 ) Int. Ci. to a new perceptual event. A metric for the subject is A61B 5 /0484 ( 2006 .01 ) determined based on the set of events . The metric is ana A61B 5 /00 ( 2006 .01 ) lyzed to determine whether the subject exhibits signs asso GO6N 20 / 10 (2006 .01 ) ciated with a condition that is associated with binocular A61B 5 /0478 ( 2006 .01 ) rivalry . -

Neurotransmitters-Drugs Andbrain Function.Pdf
Neurotransmitters, Drugs and Brain Function. Edited by Roy Webster Copyright & 2001 John Wiley & Sons Ltd ISBN: Hardback 0-471-97819-1 Paperback 0-471-98586-4 Electronic 0-470-84657-7 Neurotransmitters, Drugs and Brain Function Neurotransmitters, Drugs and Brain Function. Edited by Roy Webster Copyright & 2001 John Wiley & Sons Ltd ISBN: Hardback 0-471-97819-1 Paperback 0-471-98586-4 Electronic 0-470-84657-7 Neurotransmitters, Drugs and Brain Function Edited by R. A. Webster Department of Pharmacology, University College London, UK JOHN WILEY & SONS, LTD Chichester Á New York Á Weinheim Á Brisbane Á Singapore Á Toronto Neurotransmitters, Drugs and Brain Function. Edited by Roy Webster Copyright & 2001 John Wiley & Sons Ltd ISBN: Hardback 0-471-97819-1 Paperback 0-471-98586-4 Electronic 0-470-84657-7 Copyright # 2001 by John Wiley & Sons Ltd. Bans Lane, Chichester, West Sussex PO19 1UD, UK National 01243 779777 International ++44) 1243 779777 e-mail +for orders and customer service enquiries): [email protected] Visit our Home Page on: http://www.wiley.co.uk or http://www.wiley.com All Rights Reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording, scanning or otherwise, except under the terms of the Copyright, Designs and Patents Act 1988 or under the terms of a licence issued by the Copyright Licensing Agency Ltd, 90 Tottenham Court Road, London W1P0LP,UK, without the permission in writing of the publisher. Other Wiley Editorial Oces John Wiley & Sons, Inc., 605 Third Avenue, New York, NY 10158-0012, USA WILEY-VCH Verlag GmbH, Pappelallee 3, D-69469 Weinheim, Germany John Wiley & Sons Australia, Ltd. -

GABA Receptors
D Reviews • BIOTREND Reviews • BIOTREND Reviews • BIOTREND Reviews • BIOTREND Reviews Review No.7 / 1-2011 GABA receptors Wolfgang Froestl , CNS & Chemistry Expert, AC Immune SA, PSE Building B - EPFL, CH-1015 Lausanne, Phone: +41 21 693 91 43, FAX: +41 21 693 91 20, E-mail: [email protected] GABA Activation of the GABA A receptor leads to an influx of chloride GABA ( -aminobutyric acid; Figure 1) is the most important and ions and to a hyperpolarization of the membrane. 16 subunits with γ most abundant inhibitory neurotransmitter in the mammalian molecular weights between 50 and 65 kD have been identified brain 1,2 , where it was first discovered in 1950 3-5 . It is a small achiral so far, 6 subunits, 3 subunits, 3 subunits, and the , , α β γ δ ε θ molecule with molecular weight of 103 g/mol and high water solu - and subunits 8,9 . π bility. At 25°C one gram of water can dissolve 1.3 grams of GABA. 2 Such a hydrophilic molecule (log P = -2.13, PSA = 63.3 Å ) cannot In the meantime all GABA A receptor binding sites have been eluci - cross the blood brain barrier. It is produced in the brain by decarb- dated in great detail. The GABA site is located at the interface oxylation of L-glutamic acid by the enzyme glutamic acid decarb- between and subunits. Benzodiazepines interact with subunit α β oxylase (GAD, EC 4.1.1.15). It is a neutral amino acid with pK = combinations ( ) ( ) , which is the most abundant combi - 1 α1 2 β2 2 γ2 4.23 and pK = 10.43. -

(19) United States (12) Patent Application Publication (10) Pub
US 20130289061A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2013/0289061 A1 Bhide et al. (43) Pub. Date: Oct. 31, 2013 (54) METHODS AND COMPOSITIONS TO Publication Classi?cation PREVENT ADDICTION (51) Int. Cl. (71) Applicant: The General Hospital Corporation, A61K 31/485 (2006-01) Boston’ MA (Us) A61K 31/4458 (2006.01) (52) U.S. Cl. (72) Inventors: Pradeep G. Bhide; Peabody, MA (US); CPC """"" " A61K31/485 (201301); ‘4161223011? Jmm‘“ Zhu’ Ansm’ MA. (Us); USPC ......... .. 514/282; 514/317; 514/654; 514/618; Thomas J. Spencer; Carhsle; MA (US); 514/279 Joseph Biederman; Brookline; MA (Us) (57) ABSTRACT Disclosed herein is a method of reducing or preventing the development of aversion to a CNS stimulant in a subject (21) App1_ NO_; 13/924,815 comprising; administering a therapeutic amount of the neu rological stimulant and administering an antagonist of the kappa opioid receptor; to thereby reduce or prevent the devel - . opment of aversion to the CNS stimulant in the subject. Also (22) Flled' Jun‘ 24’ 2013 disclosed is a method of reducing or preventing the develop ment of addiction to a CNS stimulant in a subj ect; comprising; _ _ administering the CNS stimulant and administering a mu Related U‘s‘ Apphcatlon Data opioid receptor antagonist to thereby reduce or prevent the (63) Continuation of application NO 13/389,959, ?led on development of addiction to the CNS stimulant in the subject. Apt 27’ 2012’ ?led as application NO_ PCT/US2010/ Also disclosed are pharmaceutical compositions comprising 045486 on Aug' 13 2010' a central nervous system stimulant and an opioid receptor ’ antagonist. -

Mechanism of Action of Ethylenediamine On
Stone, T.W., Lui, C. and Addae, J.I. (2010) Effects of ethylenediamine – a putative GABA-releasing agent – on rat hippocampal slices and neocortical activity in vivo. European Journal of Pharmacology, 650 (2-3). pp. 568-578. ISSN 0014-2999. http://eprints.gla.ac.uk/46340/ Deposited on: 22 November 2010 Enlighten – Research publications by members of the University of Glasgow http://eprints.gla.ac.uk Effects of ethylenediamine – a putative GABA-releasing agent – on rat hippocampal slices and neocortical activity in vivo Trevor W Stone1 , Caleb Lui1 and Jonas I. Addae2 1 Neuroscience and Molecular Pharmacology, Faculty of Biomedical and Life Sciences, University of Glasgow, Glasgow G12 8QQ, U.K. 2 Dept of Preclinical Sciences, St. Augustine Campus University of the West Indies, Trinidad and Tobago. Correspondence: Prof T. W. Stone, West Medical Building, University of Glasgow, Glasgow G12 8QQ, UK. Tel - +44 141 330 4481; fax - +44 141 330 5481; e-mail - [email protected] 1 ABSTRACT The simple diamine diaminoethane (ethylenediamine, EDA) has been shown to activate GABA receptors in the central and peripheral nervous systems, partly by a direct action and partly by releasing endogenous GABA. These effects have been shown to be produced by the complexation of EDA with bicarbonate to form a carbamate. The present work has compared EDA, GABA and β-alanine responses in rat CA1 neurons using extracellular and intracellular recordings, as well as neocortical evoked potentials in vivo. Superfusion of GABA onto hippocampal slices produced depolarisation and a decrease of field epsps, both effects fading rapidly, but showing sensitivity to blockade by bicuculline. -

A Review of Glutamate Receptors I: Current Understanding of Their Biology
J Toxicol Pathol 2008; 21: 25–51 Review A Review of Glutamate Receptors I: Current Understanding of Their Biology Colin G. Rousseaux1 1Department of Pathology and Laboratory Medicine, Faculty of Medicine, University of Ottawa, Ottawa, Ontario, Canada Abstract: Seventy years ago it was discovered that glutamate is abundant in the brain and that it plays a central role in brain metabolism. However, it took the scientific community a long time to realize that glutamate also acts as a neurotransmitter. Glutamate is an amino acid and brain tissue contains as much as 5 – 15 mM glutamate per kg depending on the region, which is more than of any other amino acid. The main motivation for the ongoing research on glutamate is due to the role of glutamate in the signal transduction in the nervous systems of apparently all complex living organisms, including man. Glutamate is considered to be the major mediator of excitatory signals in the mammalian central nervous system and is involved in most aspects of normal brain function including cognition, memory and learning. In this review, the basic biology of the excitatory amino acids glutamate, glutamate receptors, GABA, and glycine will first be explored. In the second part of this review, the known pathophysiology and pathology will be described. (J Toxicol Pathol 2008; 21: 25–51) Key words: glutamate, glycine, GABA, glutamate receptors, ionotropic, metabotropic, NMDA, AMPA, review Introduction and Overview glycine), peptides (vasopressin, somatostatin, neurotensin, etc.), and monoamines (norepinephrine, dopamine and In the first decades of the 20th century, research into the serotonin) plus acetylcholine. chemical mediation of the “autonomous” (autonomic) Glutamatergic synaptic transmission in the mammalian nervous system (ANS) was an area that received much central nervous system (CNS) was slowly established over a research activity. -

Tutu Toxicity: Three Case Reports of Coriaria Arborea Ingestion, Review
THE NEW ZEALAND MEDICAL JOURNAL Journal of the New Zealand Medical Association Tutu toxicity: three case reports of Coriaria arborea ingestion, review of literature and recommendations for management Sally F Belcher, Tom R Morton Abstract We describe three cases of tutu berry (Coriaria arborea ) ingestion resulting in tonic- clonic seizures in two individuals and mild symptoms in the third. Tutu poisoning in humans appears to be a rare occurrence; the last reported case in the medical literature being over 40 years ago. We review the literature on tutu poisoning and recommend extending the period of observation for poisoned individuals from 8 hours to 12 hours or longer. We also recommend that prophylactic benzodiazepine use should be considered in those with mild to moderate symptoms of poisoning. Background There are about 30 species of Coriariaceae found around the world including southern Europe, eastern Asia, south and central America, and New Zealand.1 The six species native to New Zealand and the Chatham islands (Coriaria angustissim, C. arborea, C. lurida, C. plumosa, C. pteridoides and C. sarmentosa ) are all known by the name tutu 1 and are mostly deciduous shrubs found in grassland. There is variety in appearance and distribution as seen with Coriaria arborea, or “tree tutu”, which may become an evergreen tree growing to 6 metres in height and being found in coastal and montaine forest.2 The primary toxin, tutin , was discovered in 1870 3 and is found in all varieties of tutu 1,2 . It is a picrotoxin-like toxin which acts as an antagonist at amino acid receptors within the CNS, especially the medullary, cortical, respiratory, vasomotor, and autonomic centers 4. -

Ion Channels
UC Davis UC Davis Previously Published Works Title THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Ion channels. Permalink https://escholarship.org/uc/item/1442g5hg Journal British journal of pharmacology, 176 Suppl 1(S1) ISSN 0007-1188 Authors Alexander, Stephen PH Mathie, Alistair Peters, John A et al. Publication Date 2019-12-01 DOI 10.1111/bph.14749 License https://creativecommons.org/licenses/by/4.0/ 4.0 Peer reviewed eScholarship.org Powered by the California Digital Library University of California S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2019/20: Ion channels. British Journal of Pharmacology (2019) 176, S142–S228 THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Ion channels Stephen PH Alexander1 , Alistair Mathie2 ,JohnAPeters3 , Emma L Veale2 , Jörg Striessnig4 , Eamonn Kelly5, Jane F Armstrong6 , Elena Faccenda6 ,SimonDHarding6 ,AdamJPawson6 , Joanna L Sharman6 , Christopher Southan6 , Jamie A Davies6 and CGTP Collaborators 1School of Life Sciences, University of Nottingham Medical School, Nottingham, NG7 2UH, UK 2Medway School of Pharmacy, The Universities of Greenwich and Kent at Medway, Anson Building, Central Avenue, Chatham Maritime, Chatham, Kent, ME4 4TB, UK 3Neuroscience Division, Medical Education Institute, Ninewells Hospital and Medical School, University of Dundee, Dundee, DD1 9SY, UK 4Pharmacology and Toxicology, Institute of Pharmacy, University of Innsbruck, A-6020 Innsbruck, Austria 5School of Physiology, Pharmacology and Neuroscience, University of Bristol, Bristol, BS8 1TD, UK 6Centre for Discovery Brain Science, University of Edinburgh, Edinburgh, EH8 9XD, UK Abstract The Concise Guide to PHARMACOLOGY 2019/20 is the fourth in this series of biennial publications. The Concise Guide provides concise overviews of the key properties of nearly 1800 human drug targets with an emphasis on selective pharmacology (where available), plus links to the open access knowledgebase source of drug targets and their ligands (www.guidetopharmacology.org), which provides more detailed views of target and ligand properties. -

Interactions Between Ethanol and the Endocannabinoid System at Gabaergic Synapses on Basolateral Amygdala Principal Neurons
Alcohol 49 (2015) 781e794 Contents lists available at ScienceDirect Alcohol journal homepage: http://www.alcoholjournal.org/ Interactions between ethanol and the endocannabinoid system at GABAergic synapses on basolateral amygdala principal neurons Giuseppe Talani a,b, David M. Lovinger a,* a Section on Synaptic Pharmacology, Laboratory for Integrative Neuroscience, National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, Bethesda, MD 20892, USA b Institute of Neuroscience, National Research Council, Monserrato, Cagliari, Italy article info abstract Article history: The basolateral amygdala (BLA) plays crucial roles in stimulus value coding, as well as drug and alcohol Received 14 April 2015 dependence. Ethanol alters synaptic transmission in the BLA, while endocannabinoids (eCBs) produce Received in revised form presynaptic depression at BLA synapses. Recent studies suggest interactions between ethanol and eCBs 11 August 2015 that have important consequences for alcohol drinking behavior. To determine how ethanol and eCBs Accepted 25 August 2015 interact in the BLA, we examined the physiology and pharmacology of GABAergic synapses onto BLA pyramidal neurons in neurons from young rats. Application of ethanol at concentrations relevant to intoxication increased, in both young and adult animals, the frequency of spontaneous and miniature Keywords: Alcohol GABAergic inhibitory postsynaptic currents, indicating a presynaptic site of ethanol action. Ethanol did CB1 receptor not potentiate sIPSCs during inhibition of adenylyl cyclase while still exerting its effect during inhibition Synapse of protein kinase A. Activation of type 1 cannabinoid receptors (CB1) in the BLA inhibited GABAergic Inhibition transmission via an apparent presynaptic mechanism, and prevented ethanol potentiation. Surprisingly, Arachidonoyl ethanolamide ethanol potentiation was also prevented by CB1 antagonists/inverse agonists. -

Neurochemical and Behavioral Features in Genetic Absence Epilepsy and in Acutely Induced Absence Seizures
Hindawi Publishing Corporation ISRN Neurology Volume 2013, Article ID 875834, 48 pages http://dx.doi.org/10.1155/2013/875834 Review Article Neurochemical and Behavioral Features in Genetic Absence Epilepsy and in Acutely Induced Absence Seizures A. S. Bazyan1 and G. van Luijtelaar2 1 Institute of Higher Nervous Activity and Neurophysiology, Russian Academy of Science, Russian Federation, 5A Butlerov Street, Moscow 117485, Russia 2 Biological Psychology, Donders Centre for Cognition, Donders Institute for Brain, Cognition and Behavior, Radboud University Nijmegen, P.O. Box 9104, 6500 HE Nijmegen, The Netherlands Correspondence should be addressed to G. van Luijtelaar; [email protected] Received 21 January 2013; Accepted 6 February 2013 Academic Editors: R. L. Macdonald, Y. Wang, and E. M. Wassermann Copyright © 2013 A. S. Bazyan and G. van Luijtelaar. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. The absence epilepsy typical electroencephalographic pattern of sharp spikes and slow waves (SWDs) is considered to be dueto an interaction of an initiation site in the cortex and a resonant circuit in the thalamus. The hyperpolarization-activated cyclic nucleotide-gated cationic Ih pacemaker channels (HCN) play an important role in the enhanced cortical excitability. The role of thalamic HCN in SWD occurrence is less clear. Absence epilepsy in the WAG/Rij strain is accompanied by deficiency of the activity of dopaminergic system, which weakens the formation of an emotional positive state, causes depression-like symptoms, and counteracts learning and memory processes. -

EFFECTS of CHRONIC and ACUTE Y-HYDROXYBUTYRATE ADMINISTRATION on MASS and the RELEASE of GROWTH HORMONE and CORTICOSTERONE
EFFECTS OF CHRONIC AND ACUTE y-HYDROXYBUTYRATE ADMINISTRATION ON MASS AND THE RELEASE OF GROWTH HORMONE AND CORTICOSTERONE A Thesis Presented to the Faculty of the College of Science and Technology Morehead State University In Partial Fulfillment of the Requirements for the Degree Master of Science in Biology by Eric C. Goshorn October, 1998 /-t?p-1\'/ I"\s 4 ,he-a ·1 ~ ~lC\ .9~ G ls> 1 lo .JL. Accepted by the faculty of the College of Science and Technology, Morehead State University, in partial fulfillment of the requirements for the Master of Science degree. Master's Committee: /2..-lo -</'?, Date ii EFFECTS OF CHRONIC AND ACUTE y-HYDROXYBUTYRATE ADMINISTRATION ON MASS AND THE RELEASE OF GROWTH HORMONE AND CORTICOSTERONE Eric Christopher Goshorn, M.S. Morehead State University, 1998 Director of Thesis· Gamma-hydroxybutyrate (GHB) is a naturally occurring compound found both in neural (Roth and Suhr 1970) and extraneural tissue (Nelson et al. 1981 ). While the endogenous function of peripheral GHB has yet to be elucidated, experimentation has found that this compound induces anesthesia, and the release of growth hormone and prolactin from the anterior pituitary (Oyama and Takiguchi 1970; Takahara et al. 1977). This study was undertaken to determine the effect of chronic GHB injections on the weight gain of rats during early postnatal development. Acute injections were also administered to rats not previously treated with exogenous GHB to iii ascertain the mechanism of growth hormone release and to determine GHB's effect on corticosterone release. At the age of 3 days, both male and female rats from the chronic group were injected intraperitoneally with 100 mg/kg, 500 mg/kg, or 0 mg/kg of GHB.