Mechanism of U18666a-Mediated Cell Death in Cultured Neurons
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Integrated Bioinformatic Analysis of RNA Binding Proteins in Hepatocellular Carcinoma
www.aging-us.com AGING 2021, Vol. 13, No. 2 Research Paper Integrated bioinformatic analysis of RNA binding proteins in hepatocellular carcinoma Ling Wang1,*, Zhen Zhang2,3,*, Yuan Li1, Yanyan Wan1, Baocai Xing1,& 1Key Laboratory of Carcinogenesis and Translational Research, Ministry of Education, Hepatopancreatobiliary Surgery Department I, Peking University Cancer Hospital and Institute, Beijing 100142, China 2Department of Gastroenterological Surgery, Peking University People’s Hospital, Beijing 100044, China 3Laboratory of Surgical Oncology, Beijing Key Laboratory of Colorectal Cancer Diagnosis and Treatment Research, Peking University People’s Hospital, Beijing 100044, China *Equal contribution Correspondence to: Baocai Xing; email: [email protected] Keywords: RNA binding protein, hepatocellular carcinoma, biomarker, transcriptomics, proteomics Received: June 8, 2020 Accepted: November 3, 2020 Published: December 19, 2020 Copyright: © 2020 Wang et al. This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT RNA binding proteins (RBPs) are aberrantly expressed in a tissue-specific manner across many tumors. These proteins, which play a vital role in post-transcriptional gene regulation, are involved in RNA splicing, maturation, transport, stability, degradation, and translation. We set out to establish an accurate risk score model based on RBPs to estimate prognosis in hepatocellular carcinoma (HCC). RNA-sequencing data, proteomic data and corresponding clinical information were acquired from the Cancer Genome Atlas database and the Clinical Proteomic Tumor Analysis Consortium database respectively. We identified 406 differentially expressed RBPs between HCC tumor and normal tissues at the transcriptional and protein level. -

Predicting Clinical Response to Treatment with a Soluble Tnf-Antagonist Or Tnf, Or a Tnf Receptor Agonist
(19) TZZ _ __T (11) EP 2 192 197 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 02.06.2010 Bulletin 2010/22 C12Q 1/68 (2006.01) (21) Application number: 08170119.5 (22) Date of filing: 27.11.2008 (84) Designated Contracting States: (72) Inventor: The designation of the inventor has not AT BE BG CH CY CZ DE DK EE ES FI FR GB GR yet been filed HR HU IE IS IT LI LT LU LV MC MT NL NO PL PT RO SE SI SK TR (74) Representative: Habets, Winand Designated Extension States: Life Science Patents AL BA MK RS PO Box 5096 6130 PB Sittard (NL) (71) Applicant: Vereniging voor Christelijk Hoger Onderwijs, Wetenschappelijk Onderzoek en Patiëntenzorg 1081 HV Amsterdam (NL) (54) Predicting clinical response to treatment with a soluble tnf-antagonist or tnf, or a tnf receptor agonist (57) The invention relates to methods for predicting a clinical response to a therapy with a soluble TNF antagonist, TNF or a TNF receptor agonist and a kit for use in said methods. EP 2 192 197 A1 Printed by Jouve, 75001 PARIS (FR) EP 2 192 197 A1 Description [0001] The invention relates to methods for predicting a clinical response to a treatment with a soluble TNF antagonist, with TNF or a TNF receptor agonist using expression levels of genes of the Type I INF pathway and a kit for use in said 5 methods. In another aspect, the invention relates to a method for evaluating a pharmacological effect of a treatment with a soluble TNF antagonist, TNF or a TNF receptor agonist. -

Rrna Types in Zebrafish Development
UvA-DARE (Digital Academic Repository) Distinct maternal and somatic rRNA types in zebrafish development Locati, M. Publication date 2017 Document Version Final published version License Other Link to publication Citation for published version (APA): Locati, M. (2017). Distinct maternal and somatic rRNA types in zebrafish development. General rights It is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), other than for strictly personal, individual use, unless the work is under an open content license (like Creative Commons). Disclaimer/Complaints regulations If you believe that digital publication of certain material infringes any of your rights or (privacy) interests, please let the Library know, stating your reasons. In case of a legitimate complaint, the Library will make the material inaccessible and/or remove it from the website. Please Ask the Library: https://uba.uva.nl/en/contact, or a letter to: Library of the University of Amsterdam, Secretariat, Singel 425, 1012 WP Amsterdam, The Netherlands. You will be contacted as soon as possible. UvA-DARE is a service provided by the library of the University of Amsterdam (https://dare.uva.nl) Download date:04 Oct 2021 DISTINCT MATERNAL AND SOMATIC rRNA TYPES IN ZEBRAFISH DEVELOPMENT MAURO LOCATI DISTINCT MATERNAL AND SOMATIC rRNA TYPES IN ZEBRAFISH DEVELOPMENT MAURO LOCATI Distinct Maternal and Somatic rRNA Types in Zebrafish Development ACADEMISCH PROEFSCHRIFT ter verkrijging van de graad van doctor aan de Universiteit van Amsterdam op gezag van de Rector Magnificus prof. dr. ir. K.I.J. Maex ten overstaan van een door het College voor Promoties ingestelde commissie, in het openbaar te verdedigen in de Aula der Universiteit op woensdag 6 december 2017, te 13:00 uur door Mauro Locati geboren te Palermo, Italië Promotiecommissie: Promotor: Prof. -

Intersections Spring Symposium
INTERSECTIONS SPRING SYMPOSIUM APRIL TH, 8 Last Name First Name Project Title Page Analyzing the Effect of Urbanization on Abboud Bissan Phylogenetic signal in the Species Columba 1 Abouzahra Sharifah Peripheral IV Catheter Sterilization Device 2 Development of a Liquid Crystal Based Electron Adkins Raymond Shower Detector 2 Purification of Secreted Protein, Acidic and Rich in Cysteine (SPARC) Implicated for its Effects on Al Bahrani Zaid Intraocular Pressure 3 A Window into Metabolite Changes of Live Aljawad Yaqeen Bacteria Following Drug Infusion 3 Anderson Emily In-situ Resource Utilization on Mars 4 The Relationship Between Subjective and Objective Language Assessments For Second- Arredondo Kaitlynn Language Learners 4 Atkinson Scott MotionSense Wave: Toucheless Faucet 5 Ayyar Aneeka Hand Tremor Reduction Device 6 Modifying Low-Emissivity Glass for Use with Bailey Jessica Surface Enhanced Raman Spectroscopy 5 Automated Segmentation and Radiomic Characterization of Visceral Fat on Bowel MRIs for Barbur Iulia Crohn's Disease 6 Periodic Liquid Crystal Order: Theory and Barnaby Gavin-Rae Simulations 7 The Role of Dentistry in the U.S. Opioid Epidemic: Basore Makenna A Literature Review 7 Berends Hannah A Leader’s Role in a Nation’s Stability 8 A Study on Daily Cycles in Activity Levels of Bhatia Shireen Coyotes (Canis latrans) 8 Co-creation of an Intervention with African American Older Adults to Manage Stress Blackshire Gabrielle Associated with Self-management of 9 Developing to Developed: Entrepreneurship in Blatt Noah Latin America -

This Thesis Has Been Submitted in Fulfilment of the Requirements for a Postgraduate Degree (E.G
This thesis has been submitted in fulfilment of the requirements for a postgraduate degree (e.g. PhD, MPhil, DClinPsychol) at the University of Edinburgh. Please note the following terms and conditions of use: • This work is protected by copyright and other intellectual property rights, which are retained by the thesis author, unless otherwise stated. • A copy can be downloaded for personal non-commercial research or study, without prior permission or charge. • This thesis cannot be reproduced or quoted extensively from without first obtaining permission in writing from the author. • The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the author. • When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given. Regulation and Function of miR-199-3p in Murine and Human Cytomegalovirus Infections Nouf N. M. Laqtom s0898296 A thesis submitted in fulfilment of requirements for the degree of Doctor of Philosophy Division of Pathway Medicine, School of Biomedical Sciences The University of Edinburgh May 2013 Supervisors: Dr. Amy Buck and Dr. Bernadette Dutia i Declaration of Authorship I hereby declare that this thesis is of my own composition, and that it contains no material previously submitted for the award of any other degree. The work reported in this thesis has been executed by myself, except where due acknowledgement is made in the text. Nouf N. M. Laqtom ii Abstract Human Cytomegalovirus (HCMV), the prototypic β-herpesvirus, is the most common cause of congenital infections as well as morbidity and mortality in immunocompromised patients. -

RNA Polymerase I Transcription and Pre-Rrna Processing Are Linked by Specific SSU Processome Components
Downloaded from genesdev.cshlp.org on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press RNA polymerase I transcription and pre-rRNA processing are linked by specific SSU processome components Jennifer E.G. Gallagher,2 David A. Dunbar,3 Sander Granneman,1 Brianna M. Mitchell,3 Yvonne Osheim,4 Ann L. Beyer,4 and Susan J. Baserga1,2,3,5 1Department of Molecular Biophysics and Biochemistry, 2Department of Genetics, and 3Department of Therapeutic Radiology, Yale University School of Medicine, New Haven, Connecticut 06520-8024, USA; 4Department of Microbiology, University of Virginia, Charlottesville, Virginia 22904, USA Sequential events in macromolecular biosynthesis are often elegantly coordinated. The small ribosomal subunit (SSU) processome is a large ribonucleoprotein (RNP) required for processing of precursors to the small subunit RNA, the 18S, of the ribosome. We have found that a subcomplex of SSU processome proteins, the t-Utps, is also required for optimal rRNA transcription in vivo in the yeast Saccharomyces cerevisiae.The t-Utps are ribosomal chromatin (r-chromatin)-associated, and they exist in a complex in the absence of the U3 snoRNA. Transcription is required neither for the formation of the subcomplex nor for its r-chromatin association. The t-Utps are associated with the pre-18S rRNAs independent of the presence of the U3 snoRNA. This association may thus represent an early step in the formation of the SSU processome. Our results indicate that rRNA transcription and pre-rRNA processing are coordinated via specific components of the SSU processome. [Keywords: Ribosome; RNA; transcription; RNA processing; SSU processome] Received May 27, 2004; revised version accepted August 19, 2004. -

Expression of Distinct Maternal and Somatic 5.8S, 18S, and 28S Rrna Types During Zebrafish Development
Downloaded from rnajournal.cshlp.org on September 25, 2021 - Published by Cold Spring Harbor Laboratory Press REPORT Expression of distinct maternal and somatic 5.8S, 18S, and 28S rRNA types during zebrafish development MAURO D. LOCATI,1,3 JOHANNA F.B. PAGANO,1,3 GENEVIÈVE GIRARD,1 WIM A. ENSINK,1 MARINA VAN OLST,1 SELINA VAN LEEUWEN,1 ULRIKE NEHRDICH,2 HERMAN P. SPAINK,2 HAN RAUWERDA,1 MARTIJS J. JONKER,1 ROB J. DEKKER,1 and TIMO M. BREIT1 1RNA Biology and Applied Bioinformatics Research Group, Swammerdam Institute for Life Sciences, Faculty of Science, University of Amsterdam, Amsterdam 1090 GE, the Netherlands 2Department of Molecular Cell Biology, Institute of Biology, Leiden University, Gorlaeus Laboratories–Cell Observatorium, Leiden 2333 CE, the Netherlands ABSTRACT There is mounting evidence that the ribosome is not a static translation machinery, but a cell-specific, adaptive system. Ribosomal variations have mostly been studied at the protein level, even though the essential transcriptional functions are primarily performed by rRNAs. At the RNA level, oocyte-specific 5S rRNAs are long known for Xenopus. Recently, we described for zebrafish a similar system in which the sole maternal-type 5S rRNA present in eggs is replaced completely during embryonic development by a somatic-type. Here, we report the discovery of an analogous system for the 45S rDNA elements: 5.8S, 18S, and 28S. The maternal-type 5.8S, 18S, and 28S rRNA sequences differ substantially from those of the somatic-type, plus the maternal-type rRNAs are also replaced by the somatic-type rRNAs during embryogenesis. We discuss the structural and functional implications of the observed sequence differences with respect to the translational functions of the 5.8S, 18S, and 28S rRNA elements. -

Cryo-EM Structure and Rrna Model of a Translating Eukaryotic 80S Ribosome at 5.5-Å Resolution
Cryo-EM structure and rRNA model of a translating eukaryotic 80S ribosome at 5.5-Å resolution Jean-Paul Armachea,1, Alexander Jarascha,1, Andreas M. Angera,1, Elizabeth Villab, Thomas Beckera, Shashi Bhushana, Fabrice Jossinetc, Michael Habeckd,e, Gülcin Dindara, Sibylle Franckenberga, Viter Marqueza, Thorsten Mielkef,g, Michael Thommh, Otto Berninghausena, Birgitta Beatrixa, Johannes Södinga, Eric Westhofc, Daniel N. Wilsona,2, and Roland Beckmanna,2 aGene Center and Center for Integrated Protein Science Munich, Department of Biochemistry, Ludwig-Maximilians-Universität München, Feodor-Lynen-Strasse 25, 81377 Munich, Germany; bDepartment of Molecular Structural Biology, Max Planck Institute of Biochemistry, Am Klopferspitz 18, 82152 Martinsried, Germany; cUniversity de Strasbourg, Institut de Biologie Moleculaire et Cellulaire du Centre National de la Recherche Scientifique, 15 Rue René Descartes, 67084 Strasbourg, France; dDepartment of Empirical Inference, Max Planck Institute for Biological Cybernetics, Spemannstrasse 38, 72076 Tübingen, Germany; eDepartment of Protein Evolution, Max Planck Institute for Developmental Biology, Spemannstrasse 35, 72076 Tübingen, Germany; fUltraStrukturNetzwerk, Max Planck Institute for Molecular Genetics, Ihnestrasse 73, 14195 Berlin, Germany; gInstitut für Medizinische Physik und Biophysik, Charité, Ziegelstrasse 5-8, 10117 Berlin, Germany; and hUniversität Regensburg, Lehrstuhl für Mikrobiologie, Universitätstrasse 31, 93053 Regensburg, Germany Edited* by Gunter Blobel, The Rockefeller University, New York, NY, and approved September 8, 2010 (received for review July 9, 2010) Protein biosynthesis, the translation of the genetic code into poly- refs. 17–20. The first cryo-EM reconstruction of a eukaryotic peptides, occurs on ribonucleoprotein particles called ribosomes. 80S ribosome was reported for wheat germ (Triticum aestivum) Although X-ray structures of bacterial ribosomes are available, at 38 Å (21). -

Interaction of Rrna with Mrna and Trna in Translating Mammalian Ribosome: Functional Implications in Health and Disease
biomolecules Review Interaction of rRNA with mRNA and tRNA in Translating Mammalian Ribosome: Functional Implications in Health and Disease Amandine Bastide and Alexandre David * IGF, CNRS, INSERM, University of Montpellier, F-34094 Montpellier, France; [email protected] * Correspondence: [email protected]; Tel.: +33-4-34-359-267; Fax: +33-34-352-432 Received: 27 July 2018; Accepted: 13 September 2018; Published: 26 September 2018 Abstract: RNA-RNA interaction slowly emerges as a critical component for the smooth functioning of gene expression processes, in particular in translation where the central actor is an RNA powered molecular machine. Overall, ribosome dynamic results from sequential interactions between three main RNA species: ribosomal, transfer and messenger RNA (rRNA, tRNA and mRNA). In recent decades, special attention has been paid to the physical principles governing codon-anticodon pairing, whereas individual RNA positioning mostly relies on ribosomal RNA framework. Here, we provide a brief overview on the actual knowledge of RNA infrastructure throughout the process of translation in mammalian cells: where and how do these physical contacts occur? What are their potential roles and functions? Are they involved in disease development? What will be the main challenges ahead? Keywords: RNA-RNA interaction; mammalian ribosome; ribosomal RNA (rRNA); transfer RNA (tRNA); messenger RNA (mRNA); RNA modification; hepatitis C virus (HCV); X-linked dyskeratosis congenital 1. Introduction Proper folding of RNA through intra- and inter-molecular interactions is essential to the workings of several cellular processes (such as transcription, splicing and translation) that shape gene expression and drive cell fate. This is due to (i) the intimate relationship between RNA structure and function, and (ii) the fact that structured RNAs are central components of essential molecular machines such as the spliceosome, telomerase and ribosome. -

Studies on Mature Endothelial Cells and CD34+ Stem Cells
Exploring Mechanisms for Improvement of Cardiovascular Disease; Studies on Mature + Endothelial Cells and CD34 Stem Cells Sherin M. Bakhashab Institute of Cellular Medicine Faculty of Medical Sciences University of Newcastle A thesis submitted in fulfilment of the requirements for the degree of Doctor of Philosophy May 2015 Abstract ABSTRACT Cardiovascular disease (CVD) is the major cause of morbidity and mortality worldwide and in particular in diabetes mellitus (DM). Although metformin has been shown to reduce CVD in Type-2 DM clinical trials, the underlying cardioprotective mechanism remains unexplored. Objective: to determine the effect of metformin on mature endothelial and stem cells exposed to hypoxia, hyperglycaemia or both conditions combined. Human umbilical vein endothelial cells (HUVEC) were studied by scratch test for migration and flow cytometry for apoptosis after culture in euglycaemia or hyperglycaemia, and/or hypoxia with or without metformin. mRNA was assayed by whole transcript microarrays. Genes of interest were confirmed by quantitative real-time PCR (qRT-PCR), proteins by western blot assay or flow cytometry. Metformin increased cell survival and migration via activation of vascular endothelial growth factor (VEGF) signalling, through upregulation of matrix metalloproteinase 16, and ERK/mitogen-activated protein kinase signalling under hyperglycaemia-hypoxia. Paracrine secretion of CD34+ cells treated with euglycaemia or hyperglycaemia, and/or hypoxia with or without metformin was assessed by measuring pro-inflammatory cytokines, VEGFA, chemokine (C-X-C Motif) ligand 10 (CXCL10), tissue inhibitor of metalloproteinase 1 (TIMP1) and efficacy to promote in vitro tube formation by HUVECs. Additionally, miR-126 was evaluated in exosomes and exosome-depleted medium, which was found to be increased by metformin. -
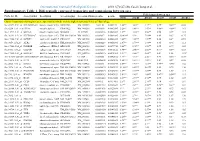
Differentially Expressed Transcripts and Comparisons Between Ages
International Journal of Biological Sciences 2010; 6(7):627-638, Caode Jiang et al. Supplementary Table 1. Differentially expressed transcripts and comparisons between ages Fold changes between ages Probe Set ID Gene Symbol Description Accession (pig) Accession (Human)p-value q-value d7/d0 d14/d0 d21/d0 d14/d7 d21/d7 d21/d14 Cluster 1:transcripts showing increased expression after birth, with the highest expression levels at 7 days of age Ssc.24673.1.S1_at HIST2H2AA4 histone cluster 2 H2aa4BX923965 NM_003516 1.22E-06 0.004472 4.88** 3.85** 3.17** 0.79* 0.65** 0.82 Ssc.6258.1.A1_a_at ITGA6 integrin alpha 6 CK463022 NM_000210 0.0000111 0.0063885 2.26** 1.49** 1.53** 0.66** 0.68** 1.03 Ssc.11712.1.A1_at KIF26A kinesin family memberBI184001 26A AL117589 0.0000126 0.0063885 3.1** 3.03** 3.06** 0.98 0.99 1.01 Ssc.26292.1.S1_at HIST1H2AC histone cluster 1 H2acXM_001928124 NM_003512 0.000017 0.0063885 10.42** 9.3* 7.01** 0.89 0.67 0.75 Ssc.17186.2.S1_at IGFBP7 insulin-like growth factorCR626227 binding proteinNM_001553 7 0.0000183 0.0063885 2.48** 2.09** 1.99** 0.84 0.8* 0.95 Ssc.3059.1.S1_at ALR2 aldo-keto reductase familyNM_001001539 1 member B1NM_003857 0.0000219 0.0066896 2.07** 1.79** 1.98** 0.86 0.96 1.11 Ssc.17186.2.S2_at POLR2B polymerase (RNA) II AF265351(DNA directed) polypeptideNM_000938 B,140kDa0.0000319 0.0077954 2.02** 1.92** 1.96** 0.95 0.97 1.02 Ssc.6435.1.S1_at COL4A1 collagen type IV alphaCN154120 1 NM_001845 0.0000339 0.0078122 3.44** 2.46** 2.05** 0.71* 0.6** 0.83 Ssc.17942.1.A1_at ELOVL7 ELOVL family memberCF176409 7 elongation -

Molecular Mechanisms Influencing the Efficiency of RNA Interference in the European Corn Borer, Ostrinia Nubilalis (Lepidoptera: Crambidae)
Molecular mechanisms influencing the efficiency of RNA interference in the European corn borer, Ostrinia nubilalis (Lepidoptera: Crambidae) by Anastasia M. W. Cooper B.S., Mississippi State University, 2010 M.A., Arkansas State University, 2014 AN ABSTRACT OF A DISSERTATION submitted in partial fulfillment of the requirements for the degree DOCTOR OF PHILOSOPHY Department of Entomology College of Agriculture KANSAS STATE UNIVERSITY Manhattan, Kansas 2020 Abstract RNA interference (RNAi) pathways function in endogenous gene regulation and protection against viruses and transposons in eukaryotic organisms. The exogenous RNAi mechanism has been utilized as a powerful reverse genetics tool to analyze gene function. In addition, RNAi- based pest management strategies are now emerging; however, applications are limited due to inefficient results for some insects, especially lepidopterans. The European corn borer (ECB), Ostrinia nubilalis, is an agriculturally relevant lepidopteran pest that exhibits very low RNAi efficiency but has a great need for new control strategies that utilize novel modes of action. This dissertation research seeks to investigate molecular mechanisms influencing RNAi efficiency in ECB and determine if commonly used reagents and tactics can improve RNAi efficiency in this species. The specific objectives were: 1) to investigate double-stranded RNA (dsRNA) stability in larval gut contents and hemolymph in ECB, 2) to identify and characterize dsRNA-degrading nuclease genes from ECB, 3) to identify and characterize core RNAi pathway genes from ECB, and 4) to compare strategies for enhancing RNAi efficiency (nanoparticles, transfection reagents, and nuclease inhibitors) in ECB. Ex vivo incubation experiments showed that dsRNA was degraded in ECB gut contents and hemolymph under physiologically relevant pH conditions.