H J2G3 (58) (685-686)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(12) Patent Application Publication (10) Pub. No.: US 2017/0020892 A1 Thompson Et Al
US 20170020892A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2017/0020892 A1 Thompson et al. (43) Pub. Date: Jan. 26, 2017 (54) USE OF NEGATIVE MODULATORS OF Related U.S. Application Data GABA RECEPTORS CONTAINING ALPHAS SUBUNITS AS FAST ACTING (60) Provisional application No. 61/972,446, filed on Mar. ANTDEPRESSANTS 31, 2014. (71) Applicant: University of Maryland, Baltimore, Publication Classification Baltimore, MD (US) (51) Int. Cl. A 6LX 3/557 (2006.01) (72) Inventors: Scott Thompson, Baltimore, MD (US); A6II 3/53 (2006.01) Mark D. Kvarta, Ellicott City, MD A6II 45/06 (2006.01) (US); Adam Van Dyke, Baltimore, MD (52) U.S. Cl. (US) CPC ........... A61 K3I/55.17 (2013.01); A61K 45/06 (2013.01); A61 K3I/53 (2013.01) (73) Assignee: University of Maryland, Baltimore, Baltimore, MD (US) (57) ABSTRACT Embodiments of the disclosure include methods and com (21) Appl. No.: 15/300,984 positions related to treatment of one or more medical conditions with one or more negative modulators of GABA (22) PCT Filed: Mar. 31, 2015 receptors. In specific embodiments, depression and/or Sui cidability is treated or ameliorated or prevented with one or (86) PCT No.: PCT/US2O15/023667 more negative modulators of GABA receptors, such as a S 371 (c)(1), partial inverse agonist of a GABA receptor comprising an (2) Date: Sep. 30, 2016 alpha5 subunit. Patent Application Publication Jan. 26, 2017. Sheet 1 of 12 US 2017/002O892 A1 ×1/ /|\ Patent Application Publication Jan. 26, 2017. Sheet 3 of 12 US 2017/002O892 A1 & Patent Application Publication Jan. -
ANNNNNNNNNNNNNNNNNNNN 100A 006 Left Eye Input Right Eye Input
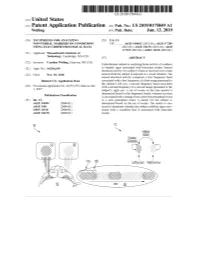
US 20190175049A1 ( 19) United States (12 ) Patent Application Publication (10 ) Pub. No. : US 2019 /0175049 A1 Welling ( 43 ) Pub . Date : Jun . 13 , 2019 ( 54 ) TECHNIQUES FOR ANALYZING (52 ) U . S . CI. NON -VERBAL MARKERS OF CONDITIONS CPC . .. A61B 5 /04842 (2013 . 01 ) ; A61B 5 / 7289 USING ELECTROPHYSIOLOGICAL DATA (2013 . 01) ; A61B 5 /0478 ( 2013 .01 ) ; A61B 5 /7225 ( 2013. 01 ) ; G06N 20 / 10 (2019 .01 ) (71 ) Applicant: Massachusetts Institute of Technology , Cambridge , MA (US ) ( 57 ) ABSTRACT (72 ) Inventor : Caroline Welling, Hanover, NH (US ) Embodiments related to analyzing brain activity of a subject to identify signs associated with binocular rivalry . Sensed ( 21 ) Appl. No. : 16 / 206, 639 electrical activity of a subject' s brain is received over a time period while the subject is exposed to a visual stimulus. The ( 22 ) Filed : Nov. 30 , 2018 sensed electrical activity comprises a first frequency band Related U . S . Application Data associated with a first frequency of a first image presented to the subject ' s left eye , a second frequency band associated (60 ) Provisional application No .62 / 593 , 535, filed on Dec . with a second frequency of a second image presented to the 1 , 2017 subject ' s right eye . A set of events in the time period is determined based on the frequency bands, wherein an event Publication Classification is associated with a change from a previous perceptual event (51 ) Int. Ci. to a new perceptual event. A metric for the subject is A61B 5 /0484 ( 2006 .01 ) determined based on the set of events . The metric is ana A61B 5 /00 ( 2006 .01 ) lyzed to determine whether the subject exhibits signs asso GO6N 20 / 10 (2006 .01 ) ciated with a condition that is associated with binocular A61B 5 /0478 ( 2006 .01 ) rivalry . -

(12) Patent Application Publication (10) Pub. No.: US 2006/0110428A1 De Juan Et Al
US 200601 10428A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2006/0110428A1 de Juan et al. (43) Pub. Date: May 25, 2006 (54) METHODS AND DEVICES FOR THE Publication Classification TREATMENT OF OCULAR CONDITIONS (51) Int. Cl. (76) Inventors: Eugene de Juan, LaCanada, CA (US); A6F 2/00 (2006.01) Signe E. Varner, Los Angeles, CA (52) U.S. Cl. .............................................................. 424/427 (US); Laurie R. Lawin, New Brighton, MN (US) (57) ABSTRACT Correspondence Address: Featured is a method for instilling one or more bioactive SCOTT PRIBNOW agents into ocular tissue within an eye of a patient for the Kagan Binder, PLLC treatment of an ocular condition, the method comprising Suite 200 concurrently using at least two of the following bioactive 221 Main Street North agent delivery methods (A)-(C): Stillwater, MN 55082 (US) (A) implanting a Sustained release delivery device com (21) Appl. No.: 11/175,850 prising one or more bioactive agents in a posterior region of the eye so that it delivers the one or more (22) Filed: Jul. 5, 2005 bioactive agents into the vitreous humor of the eye; (B) instilling (e.g., injecting or implanting) one or more Related U.S. Application Data bioactive agents Subretinally; and (60) Provisional application No. 60/585,236, filed on Jul. (C) instilling (e.g., injecting or delivering by ocular ion 2, 2004. Provisional application No. 60/669,701, filed tophoresis) one or more bioactive agents into the Vit on Apr. 8, 2005. reous humor of the eye. Patent Application Publication May 25, 2006 Sheet 1 of 22 US 2006/0110428A1 R 2 2 C.6 Fig. -

Effect of Repeated Gaboxadol Administration on Night Sleep and Next-Day Performance in Healthy Elderly Subjects
Neuropsychopharmacology (2005) 30, 833–841 & 2005 Nature Publishing Group All rights reserved 0893-133X/05 $30.00 www.neuropsychopharmacology.org Effect of Repeated Gaboxadol Administration on Night Sleep and Next-Day Performance in Healthy Elderly Subjects 1 2 ,3 1 Stefan Mathias , Josef Zihl , Axel Steiger* and Marike Lancel 1Section of Sleep Pharmacology, Max-Planck-Institute of Psychiatry, Munich, Germany; 2Section of Neuropsychology, Max-Planck-Institute of Psychiatry, Munich, Germany; 3Department of Psychiatry, Max-Planck-Institute of Psychiatry, Munich, Germany Aging is associated with dramatic reductions in sleep continuity and sleep intensity. Since gaboxadol, a selective GABAA receptor agonist, has been demonstrated to improve sleep consolidation and promote deep sleep, it may be an effective hypnotic, particularly for elderly patients with insomnia. In the present study, we investigated the effects of subchronic gaboxadol administration on nocturnal sleep and its residual effects during the next days in elderly subjects. This was a randomized, double-blind, placebo-controlled, balanced crossover study in 10 healthy elderly subjects without sleep complaints. The subjects were administered either placebo or 15 mg gaboxadol hydrochloride at bedtime on three consecutive nights. Sleep was recorded during each night from 2300 to 0700 h and tests assessing attention (target detection, stroop test) and memory function (visual form recognition, immediate word recall, digit span) were applied at 0900, 1400, and 1700 h during the following days. Compared with placebo, gaboxadol significantly shortened subjective sleep onset latency and increased self-rated sleep intensity and quality. Polysomnographic recordings showed that it significantly decreased the number of awakenings, the amount of intermittent wakefulness, and stage 1, and increased slow wave sleep and stage 2. -

GABA Receptors
D Reviews • BIOTREND Reviews • BIOTREND Reviews • BIOTREND Reviews • BIOTREND Reviews Review No.7 / 1-2011 GABA receptors Wolfgang Froestl , CNS & Chemistry Expert, AC Immune SA, PSE Building B - EPFL, CH-1015 Lausanne, Phone: +41 21 693 91 43, FAX: +41 21 693 91 20, E-mail: [email protected] GABA Activation of the GABA A receptor leads to an influx of chloride GABA ( -aminobutyric acid; Figure 1) is the most important and ions and to a hyperpolarization of the membrane. 16 subunits with γ most abundant inhibitory neurotransmitter in the mammalian molecular weights between 50 and 65 kD have been identified brain 1,2 , where it was first discovered in 1950 3-5 . It is a small achiral so far, 6 subunits, 3 subunits, 3 subunits, and the , , α β γ δ ε θ molecule with molecular weight of 103 g/mol and high water solu - and subunits 8,9 . π bility. At 25°C one gram of water can dissolve 1.3 grams of GABA. 2 Such a hydrophilic molecule (log P = -2.13, PSA = 63.3 Å ) cannot In the meantime all GABA A receptor binding sites have been eluci - cross the blood brain barrier. It is produced in the brain by decarb- dated in great detail. The GABA site is located at the interface oxylation of L-glutamic acid by the enzyme glutamic acid decarb- between and subunits. Benzodiazepines interact with subunit α β oxylase (GAD, EC 4.1.1.15). It is a neutral amino acid with pK = combinations ( ) ( ) , which is the most abundant combi - 1 α1 2 β2 2 γ2 4.23 and pK = 10.43. -

Gaboxadol Normalizes Behavioral Abnormalities in a Mouse Model of Fragile X Syndrome
ORIGINAL RESEARCH published: 25 June 2019 doi: 10.3389/fnbeh.2019.00141 Gaboxadol Normalizes Behavioral Abnormalities in a Mouse Model of Fragile X Syndrome Patricia Cogram 1,2,3,4, Robert M. J. Deacon 1,2,3,4, Jennifer L. Warner-Schmidt 5, Melanie J. von Schimmelmann 6, Brett S. Abrahams 6,7 and Matthew J. During 6,8* 1FRAXA-DVI, FRAXA Research Foundation, Boston, MA, United States, 2Centre for Systems Biotechnology, Biomedicine Division, Fraunhofer-Gesellschaft, Santiago, Chile, 3GEN.DDI Limited, London, United Kingdom, 4Institute of Ecology and Biodiversity (IEB), University of Chile, Santiago, Chile, 5NeuroJenic Consulting, LLC, Garden City, NY, United States, 6Ovid Therapeutics, New York, NY, United States, 7Department of Genetics and Neuroscience, Albert Einstein College of Medicine, Bronx, NY, United States, 8Department of Neurological Surgery and Molecular Virology, Immunology and Medical Genetics, Ohio State University College of Medicine, Columbus, OH, United States Fragile X syndrome (FXS) is the most common inherited form of intellectual disability and autism. FXS is also accompanied by attention problems, hyperactivity, anxiety, aggression, poor sleep, repetitive behaviors, and self-injury. Recent work supports the role of g-aminobutyric-acid (GABA), the primary inhibitory neurotransmitter in the brain, in mediating symptoms of FXS. Deficits in GABA machinery have been observed in a mouse model of FXS, including a loss of tonic inhibition in the amygdala, which is Edited by: Martine Ammassari-Teule, mediated by extrasynaptic GABAA receptors. Humans with FXS also show reduced Italian National Research Council GABAA receptor availability. Here, we sought to evaluate the potential of gaboxadol (CNR), Italy (also called OV101 and THIP), a selective and potent agonist for delta-subunit-containing Reviewed by: extrasynaptic GABA receptors (dSEGA), as a therapeutic agent for FXS by assessing Giulia Poggi, A University of Zurich, Switzerland its ability to normalize aberrant behaviors in a relatively uncharacterized mouse model Valerie J. -

Ligand-Gated Ion Channels' British Journal of Pharmacology, Vol
Edinburgh Research Explorer The Concise Guide to PHARMACOLOGY 2015/16 Citation for published version: Alexander, SP, Peters, JA, Kelly, E, Marrion, N, Benson, HE, Faccenda, E, Pawson, AJ, Sharman, JL, Southan, C, Davies, JA & CGTP Collaborators 2015, 'The Concise Guide to PHARMACOLOGY 2015/16: Ligand-gated ion channels' British Journal of Pharmacology, vol. 172, no. 24, pp. 5870-5903. DOI: 10.1111/bph.13350 Digital Object Identifier (DOI): 10.1111/bph.13350 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: British Journal of Pharmacology General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 05. Apr. 2019 S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2015/16: Ligand-gated ion channels. British Journal of Pharmacology (2015) 172, 5870–5903 THE CONCISE GUIDE TO PHARMACOLOGY 2015/16: Ligand-gated ion channels Stephen PH Alexander1, -

)&F1y3x PHARMACEUTICAL APPENDIX to THE
)&f1y3X PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE )&f1y3X PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 3 Table 1. This table enumerates products described by International Non-proprietary Names (INN) which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service (CAS) registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. Product CAS No. Product CAS No. ABAMECTIN 65195-55-3 ACTODIGIN 36983-69-4 ABANOQUIL 90402-40-7 ADAFENOXATE 82168-26-1 ABCIXIMAB 143653-53-6 ADAMEXINE 54785-02-3 ABECARNIL 111841-85-1 ADAPALENE 106685-40-9 ABITESARTAN 137882-98-5 ADAPROLOL 101479-70-3 ABLUKAST 96566-25-5 ADATANSERIN 127266-56-2 ABUNIDAZOLE 91017-58-2 ADEFOVIR 106941-25-7 ACADESINE 2627-69-2 ADELMIDROL 1675-66-7 ACAMPROSATE 77337-76-9 ADEMETIONINE 17176-17-9 ACAPRAZINE 55485-20-6 ADENOSINE PHOSPHATE 61-19-8 ACARBOSE 56180-94-0 ADIBENDAN 100510-33-6 ACEBROCHOL 514-50-1 ADICILLIN 525-94-0 ACEBURIC ACID 26976-72-7 ADIMOLOL 78459-19-5 ACEBUTOLOL 37517-30-9 ADINAZOLAM 37115-32-5 ACECAINIDE 32795-44-1 ADIPHENINE 64-95-9 ACECARBROMAL 77-66-7 ADIPIODONE 606-17-7 ACECLIDINE 827-61-2 ADITEREN 56066-19-4 ACECLOFENAC 89796-99-6 ADITOPRIM 56066-63-8 ACEDAPSONE 77-46-3 ADOSOPINE 88124-26-9 ACEDIASULFONE SODIUM 127-60-6 ADOZELESIN 110314-48-2 ACEDOBEN 556-08-1 ADRAFINIL 63547-13-7 ACEFLURANOL 80595-73-9 ADRENALONE -

ICADTS \(28-30 Août 2007, Seattle, USA\)
Annales de Toxicologie Analytique, vol. XIX, n° 4, 2007 ORAL PRESENTATIONS RESULTS: There were 7,673 external cause deaths in Victoria between July 2000 and June 2005. Of these, COMMUNICATIONS ORALES there were 2,086 (27%) external cause deaths contained a toxicology report where alcohol was identified as positive (greater than 0). A toxicology report was not Postmortem toxicology attached in 1,420 (18.5%) cases and the remaining Toxicologie postmortem 4,152 external cause cases contained a negative result for alcohol. Of cases with a toxicology report 455 Understanding forensic toxicology in (5.4%) had a blood alcohol concentration (BAC) of less than 0.06 g/100 mL. There were 564 (7.4%) detected relation to external-cause deaths with alcohol (0.06 g/100mL to <0.16 g/100mL) and 659 J. KILLIAN(1), O. DRUMMER(2), J. OZANNE- (8.6%) had a toxic level of alcohol (>0.16 g/100 mL). SMITH(1) Of all causes with alcohol detected, 2726 (37%) were (1) Monash University Accident Research Centre, due to intentional self-harm, followed by 2005 (27%) being transport related, 932 (12%) due to poisoning and Victoria, Australia 553 (7%) being fall related. The trends and themes of (2) Monash University Department of Forensic other drugs will also be reported especially in relation Medicine and Victorian Institute of Forensic Medicine, to traffic injuries. Victoria, Australia CONCLUSIONS: The study results provide, for the AIMS: Injuries are not only recognised as an important first time in Australia, a systematic examination of the public health problem, but are also one of the major epidemiology of licit and illicit drugs in injury deaths causes of death. -

GABA Site Agonist Gaboxadol Induces Addiction-Predicting Persistent Changes in Ventral Tegmental Area Dopamine Neurons but Is Not Rewarding in Mice Or Baboons
5310 • The Journal of Neuroscience, April 11, 2012 • 32(15):5310–5320 Behavioral/Systems/Cognitive GABA Site Agonist Gaboxadol Induces Addiction-Predicting Persistent Changes in Ventral Tegmental Area Dopamine Neurons But Is Not Rewarding in Mice or Baboons Elena Vashchinkina,1 Anne Panhelainen,1 Olga Yu. Vekovischeva,1 Teemu Aitta-aho,1 Bjarke Ebert,2 Nancy A. Ator,3 and Esa R. Korpi1 1Institute of Biomedicine, Pharmacology, University of Helsinki, FI-00014 Helsinki, Finland, 2H. Lundbeck A/S, DK-2500 Copenhagen, Denmark, and 3Division of Behavioral Biology, Psychiatry, and Behavioral Sciences, The Johns Hopkins School of Medicine, Baltimore, Maryland 21224-6823 Dopamine neurons of the ventral tegmental area (VTA) are involved at early phases of drug addiction. Even the first in vivo dose of various abused drugs induces glutamate receptor plasticity at the excitatory synapses of these neurons. Benzodiazepines that suppress the inhibitory GABAergic interneurons in the VTA via facilitation of synaptic GABAA receptors have induced neuroplas- ticity in dopamine neurons due to this disinhibitory mechanism. Here, we have tested a non-benzodiazepine direct GABA site agonist 4,5,6,7-tetrahydroisoxazolol[4,5-c]pyridine-3-ol (THIP) (also known as gaboxadol) that acts preferentially via high- affinity extrasynaptic GABAA receptors. A single sedative dose of THIP (6 mg/kg) to mice induced glutamate receptor plasticity for at least 6 d after administration. Increased AMPA/NMDA receptor current ratio and increased frequency, amplitude, and rectifi- cation of AMPA receptor responses suggested persistent targeting of GluA2-lacking AMPA receptors in excitatory synapses of VTA ␦Ϫ/Ϫ dopamine neurons ex vivo after THIP administration. -

Neurochemical and Behavioral Features in Genetic Absence Epilepsy and in Acutely Induced Absence Seizures
Hindawi Publishing Corporation ISRN Neurology Volume 2013, Article ID 875834, 48 pages http://dx.doi.org/10.1155/2013/875834 Review Article Neurochemical and Behavioral Features in Genetic Absence Epilepsy and in Acutely Induced Absence Seizures A. S. Bazyan1 and G. van Luijtelaar2 1 Institute of Higher Nervous Activity and Neurophysiology, Russian Academy of Science, Russian Federation, 5A Butlerov Street, Moscow 117485, Russia 2 Biological Psychology, Donders Centre for Cognition, Donders Institute for Brain, Cognition and Behavior, Radboud University Nijmegen, P.O. Box 9104, 6500 HE Nijmegen, The Netherlands Correspondence should be addressed to G. van Luijtelaar; [email protected] Received 21 January 2013; Accepted 6 February 2013 Academic Editors: R. L. Macdonald, Y. Wang, and E. M. Wassermann Copyright © 2013 A. S. Bazyan and G. van Luijtelaar. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. The absence epilepsy typical electroencephalographic pattern of sharp spikes and slow waves (SWDs) is considered to be dueto an interaction of an initiation site in the cortex and a resonant circuit in the thalamus. The hyperpolarization-activated cyclic nucleotide-gated cationic Ih pacemaker channels (HCN) play an important role in the enhanced cortical excitability. The role of thalamic HCN in SWD occurrence is less clear. Absence epilepsy in the WAG/Rij strain is accompanied by deficiency of the activity of dopaminergic system, which weakens the formation of an emotional positive state, causes depression-like symptoms, and counteracts learning and memory processes. -

Pharmaceutical Appendix to the Tariff Schedule 2
Harmonized Tariff Schedule of the United States (2007) (Rev. 2) Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE Harmonized Tariff Schedule of the United States (2007) (Rev. 2) Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 2 Table 1. This table enumerates products described by International Non-proprietary Names (INN) which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service (CAS) registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. ABACAVIR 136470-78-5 ACIDUM LIDADRONICUM 63132-38-7 ABAFUNGIN 129639-79-8 ACIDUM SALCAPROZICUM 183990-46-7 ABAMECTIN 65195-55-3 ACIDUM SALCLOBUZICUM 387825-03-8 ABANOQUIL 90402-40-7 ACIFRAN 72420-38-3 ABAPERIDONUM 183849-43-6 ACIPIMOX 51037-30-0 ABARELIX 183552-38-7 ACITAZANOLAST 114607-46-4 ABATACEPTUM 332348-12-6 ACITEMATE 101197-99-3 ABCIXIMAB 143653-53-6 ACITRETIN 55079-83-9 ABECARNIL 111841-85-1 ACIVICIN 42228-92-2 ABETIMUSUM 167362-48-3 ACLANTATE 39633-62-0 ABIRATERONE 154229-19-3 ACLARUBICIN 57576-44-0 ABITESARTAN 137882-98-5 ACLATONIUM NAPADISILATE 55077-30-0 ABLUKAST 96566-25-5 ACODAZOLE 79152-85-5 ABRINEURINUM 178535-93-8 ACOLBIFENUM 182167-02-8 ABUNIDAZOLE 91017-58-2 ACONIAZIDE 13410-86-1 ACADESINE 2627-69-2 ACOTIAMIDUM 185106-16-5 ACAMPROSATE 77337-76-9