Synthesis and Characterization of TPGS–Gemcitabine Prodrug
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Recommended Methods for the Identification and Analysis Of
Vienna International Centre, P.O. Box 500, 1400 Vienna, Austria Tel: (+43-1) 26060-0, Fax: (+43-1) 26060-5866, www.unodc.org RECOMMENDED METHODS FOR THE IDENTIFICATION AND ANALYSIS OF AMPHETAMINE, METHAMPHETAMINE AND THEIR RING-SUBSTITUTED ANALOGUES IN SEIZED MATERIALS (revised and updated) MANUAL FOR USE BY NATIONAL DRUG TESTING LABORATORIES Laboratory and Scientific Section United Nations Office on Drugs and Crime Vienna RECOMMENDED METHODS FOR THE IDENTIFICATION AND ANALYSIS OF AMPHETAMINE, METHAMPHETAMINE AND THEIR RING-SUBSTITUTED ANALOGUES IN SEIZED MATERIALS (revised and updated) MANUAL FOR USE BY NATIONAL DRUG TESTING LABORATORIES UNITED NATIONS New York, 2006 Note Mention of company names and commercial products does not imply the endorse- ment of the United Nations. This publication has not been formally edited. ST/NAR/34 UNITED NATIONS PUBLICATION Sales No. E.06.XI.1 ISBN 92-1-148208-9 Acknowledgements UNODC’s Laboratory and Scientific Section wishes to express its thanks to the experts who participated in the Consultative Meeting on “The Review of Methods for the Identification and Analysis of Amphetamine-type Stimulants (ATS) and Their Ring-substituted Analogues in Seized Material” for their contribution to the contents of this manual. Ms. Rosa Alis Rodríguez, Laboratorio de Drogas y Sanidad de Baleares, Palma de Mallorca, Spain Dr. Hans Bergkvist, SKL—National Laboratory of Forensic Science, Linköping, Sweden Ms. Warank Boonchuay, Division of Narcotics Analysis, Department of Medical Sciences, Ministry of Public Health, Nonthaburi, Thailand Dr. Rainer Dahlenburg, Bundeskriminalamt/KT34, Wiesbaden, Germany Mr. Adrian V. Kemmenoe, The Forensic Science Service, Birmingham Laboratory, Birmingham, United Kingdom Dr. Tohru Kishi, National Research Institute of Police Science, Chiba, Japan Dr. -

Enhancing Transdermal Delivery of Opioid Antagonists and Agonists Using Codrugs Linked to Bupropion Or Hydroxybupropion Audra L
University of Kentucky UKnowledge Pharmaceutical Sciences Faculty Patents Pharmaceutical Sciences 2-18-2014 Enhancing Transdermal Delivery of Opioid Antagonists and Agonists Using Codrugs Linked To Bupropion or Hydroxybupropion Audra L. Stinchcomb University of Kentucky Peter A. Crooks University of Kentucky, [email protected] Mohamad O. Hamad University of Kentucky, [email protected] Paul K. Kiptoo University of Kentucky Click here to let us know how access to this document benefits oy u. Follow this and additional works at: https://uknowledge.uky.edu/ps_patents Part of the Pharmacy and Pharmaceutical Sciences Commons Recommended Citation Stinchcomb, Audra L.; Crooks, Peter A.; Hamad, Mohamad O.; and Kiptoo, Paul K., "Enhancing Transdermal Delivery of Opioid Antagonists and Agonists Using Codrugs Linked To Bupropion or Hydroxybupropion" (2014). Pharmaceutical Sciences Faculty Patents. 27. https://uknowledge.uky.edu/ps_patents/27 This Patent is brought to you for free and open access by the Pharmaceutical Sciences at UKnowledge. It has been accepted for inclusion in Pharmaceutical Sciences Faculty Patents by an authorized administrator of UKnowledge. For more information, please contact [email protected]. US008653271B2 (12) United States Patent (10) Patent N0.: US 8,653,271 B2 Stinchcomb et a]. (45) Date of Patent: Feb. 18, 2014 (54) ENHANCING TRANSDERMAL DELIVERY (58) Field of Classi?cation Search OF OPIOID ANTAGONISTS AND AGONISTS USPC ............................... .. 546/45, 44, 46; 514/282 USING CODRUGS LINKED TO BUPROPION See application ?le for complete search history. OR HYDROXYBUPROPION (56) References Cited (75) Inventors: Audra L. Stinchcomb, Lexington, KY (US); Peter A. Crooks, Nicholasville, U.S. PATENT DOCUMENTS KY (US); Mohamed O. Hamad, Lexington, KY (US); Paul K. -

Detection and Characterization of Pyromarkers of Smoked Drugs of Abuse
Graduate Theses, Dissertations, and Problem Reports 2012 Detection and characterization of pyromarkers of smoked drugs of abuse Rona Kiyomi Nishikawa West Virginia University Follow this and additional works at: https://researchrepository.wvu.edu/etd Recommended Citation Nishikawa, Rona Kiyomi, "Detection and characterization of pyromarkers of smoked drugs of abuse" (2012). Graduate Theses, Dissertations, and Problem Reports. 4904. https://researchrepository.wvu.edu/etd/4904 This Dissertation is protected by copyright and/or related rights. It has been brought to you by the The Research Repository @ WVU with permission from the rights-holder(s). You are free to use this Dissertation in any way that is permitted by the copyright and related rights legislation that applies to your use. For other uses you must obtain permission from the rights-holder(s) directly, unless additional rights are indicated by a Creative Commons license in the record and/ or on the work itself. This Dissertation has been accepted for inclusion in WVU Graduate Theses, Dissertations, and Problem Reports collection by an authorized administrator of The Research Repository @ WVU. For more information, please contact [email protected]. DETECTION AND CHARACTERIZATION OF PYROMARKERS OF SMOKED DRUGS OF ABUSE Rona Kiyomi Nishikawa Dissertation submitted to the Eberly College of Arts and Sciences at West Virginia University in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Chemistry Committee of Advisors: Suzanne C. Bell, Ph.D., Chair R. Lloyd Carroll, Ph.D. Patrick S. Callery, Ph.D. Harry O. Finklea, Ph.D. Keith B. Morris, Ph.D. C. Eugene Bennett Department of Chemistry Morgantown, West Virginia 2012 Keywords: Fentanyl, Heroin, Pyrolysis, Pyromarkers, GC/MS Copyright © 2012 Rona Kiyomi Nishikawa 1 Abstract DETECTION AND CHARACTERIZATION OF PYROMARKERS OF SMOKED DRUGS OF ABUSE Rona Kiyomi Nishikawa Smoking or inhalation has increased in popularity as the choice route of administration of drugs of abuse amongst drug abusers. -

Claire Cole, Lisa Jones, Jim Mcveigh, Andrew Kicman, Qutub Syed & Mark A
Claire Cole, Lisa Jones, Jim McVeigh, Andrew Kicman, Qutub Syed & Mark A. Bellis Acknowledgements The authors would like to thank the following for their contribution to the literature searching, design, proofing, editing and distribution of this document: Carol Chadwick. Sian Connolly, Layla English, Michael Evans-Brown, Dave Seddon, Olivia Wooding, Lee Tisdall, Ellie McCoy and Gemma Parry of the Centre for Public Health. We would also like to thank the International Harm Reduction Association (IHRA) for their support with launching this report at their 2010 conference. About the Authors Claire Cole and Lisa Jones are Senior Researchers at the Centre for Public Health, Liverpool John Moores University. Jim McVeigh is the Deputy Director and Reader in Substance Use Epidemiology at the Centre for Public Health. Professor Andrew Kicman is Head of Research and Development in the Department of Forensic Science and Drug Monitoring, The Drug Control Centre, at Kings College London. Professor Qutub Syed is the Regional Director for Health Protection Agency in the North West Region. Professor Mark A. Bellis is the Director of the Centre for Public Health. 1 Contents Executive Summary 4 1. Introduction 10 2. Methodology 13 3. Heroin 17 4. Cocaine and Crack Cocaine 25 5. Amphetamine and Methamphetamine 30 6. Ecstasy 33 7. Cannabis 36 8. Other Illicit Drugs 38 9. Public Health Response and Harm Reduction Messages 40 10. Summary 45 References 47 Appendix 1 - Glossary 55 List of Tables Table 1: Summary of drug adulteration evidence 5 Table 2: Summary -

Prodrug Derivatives of Carboxylic Acid Drugs
Patentamt JEuropaischesEuropean Patent Office Office europeen des brevets © Publication number: 0 278 977 B1 © EUROPEAN PATENT SPECIFICATION © Date of publication of patent specification : © Int. CI.5 : C07C 233/01, A61 K 31/21 06.05.92 Bulletin 92/19 © Application number : 87905946.7 @ Date of filing : 25.08.87 © International application number : PCT/DK87/00104 @ International publication number : WO 88/01615 10.03.88 Gazette 88/06 (54) PRODRUG DERIVATIVES OF CARBOXYLIC ACID DRUGS. The file contains technical information (56) References cited : submitted after the application was filed and US-A- 4 588 525 not included in this specification US-A- 4 678 806 CHEMICAL ABSTRACTS, vol. 106, 1987, ab- stract no. 781 87t, 16 March 1987; H. © 26.08.86 DK 4066/86 Priority : BUNDGAARD et al, Esters of N,N-disub- stituted2-hydroxyacetamides as a novel high- © Date of publication of application : ly biolabile prodrug type for carboxylic acid 24.08.88 Bulletin 88/34 agents'' © Publication of the grant of the patent : © Proprietor : Bundgaard, Hans 06.05.92 Bulletin 92/19 36, Tjornevej DK-2970 Horsholm (DK) Niels Mork States Proprietor : NIELSEN, © Designated Contracting : st.th. AT BE CH DE FR GB IT LI LU NL SE Cumberlandsgade 15, DK-2300 Kobenhavn S (DK) References cited 56 : Inventor Hans EP-A- 0 073 397 © : Bundgaard, EP-A- 077 720 36, Tjornevej DK-2970 Horsholm EP-A- 106 541 (DK) Inventor : NIELSEN, Niels Mork EP-A- 201 829 st.th. EP-A- 224 178 Cumberlandsgade 15, DK-2300 Kobenhavn S EP-A- 227 355 (DK) EP-A- 237 051 WO-A-86/00066 © Representative : Andersen, Henrik Rastrup et CH-A- 513 815 al US-A- 4 206 220 c/o Plougmann & Vingtoft Sankt Annae Plads US-A- 4 235 887 11 P.O. -
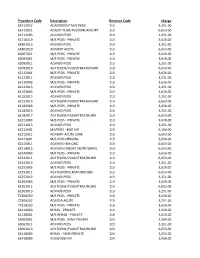
Procedure Code
Procedure Code Description Revenue Code Charge 61413012 ADJUSTMENT M/S PEDS 113 3,251.00 61413021 ADJUST TELM/PULM/NURO/ORT 113 6,653.00 61713103 ADJ M/S PEDS 113 3,251.00 61716119 M/S PEDS - PRIVATE 113 3,626.00 61801013 ADJ M/S PEDS 113 3,251.00 61802019 ADJ BMT ACUTE 113 6,653.00 62067021 M/S PEDS - PRIVATE 113 3,626.00 62093001 M/S PEDS - PRIVATE 113 3,626.00 62093011 ADJ M/S PEDS 113 3,251.00 62093013 ADJ TELEM/PULM/TRACH/NURO 113 6,653.00 62123001 M/S PEDS - PRIVATE 113 3,626.00 62123011 ADJ M/S PEDS 113 3,251.00 62133003 M/S PEDS - PRIVATE 113 3,626.00 62133013 ADJ M/S PEDS 113 3,251.00 62153003 M/S PEDS - PRIVATE 113 3,626.00 62153013 ADJ M/S PEDS 113 3,251.00 62153015 ADJ TELEM/PULM/TRACH/NURO 113 6,653.00 62183003 M/S PEDS - PRIVATE 113 3,626.00 62183013 ADJ M/S PEDS 113 3,251.00 62183017 ADJ TELEM/PULM/TRAC/NURO 113 6,653.00 62211003 M/S PEDS - PRIVATE 113 3,626.00 62211015 ADJ M/S PEDS 113 3,251.00 62212001 MS/PEDS - BMT HIP 113 6,160.00 62212011 ADJ BMT-ACUTE CARE 113 6,653.00 62213001 M/S PEDS HEMONC 113 3,626.00 62213011 ADJ M/S HEM ONC 113 6,653.00 62214015 ADJ M/S CARDIAC MONITORNG 113 6,653.00 62243003 M/S PEDS - PRIVATE 113 3,626.00 62243011 ADJ TELEM/PULM/TRAC/NURO 113 6,653.00 62243013 ADJ M/S PEDS 113 3,251.00 62253003 M/S PEDS - PRIVATE 113 3,626.00 62253011 ADJ TELM/PULM/NURO/OBS 113 6,653.00 62253013 ADJ M/S PEDS 113 3,251.00 62263003 M/S PEDS - PRIVATE 113 3,626.00 62263011 ADJ TELEM/PULM/TRAC/NURO 113 6,653.00 62263013 ADJ M/S PEDS 113 3,251.00 72309250 M/S PEDS - PRIVATE 113 3,626.00 72309252 ADJ EDA ACUTE 113 -

ED 042 219 DOCUMENT RESUME CG 005 800 the Development of A
DOCUMENT RESUME ED 042 219 24 CG 005 800 TITLE The Development of a Curriculum for Teaching Elementary and Secondary School Children the Dangers Inherent in the Use and Abuse of Dangerous Drugs. Final Progress Report. INSTITUTION Laredo Independent School District, Tex. SPONS AGENCY Office of Education (DHEW) ,Washington, D.C. BUREATI NO BR-9-G-067 PUB DATE 30 Sep 70 CONTRACT OEC -7 -9- 530067 -0123- (010) NOTE 545p.; Second Edition EDRS PRICE EDRS Price MF-$2.00 HC-$27.35 DESCRIPTORS *Curriculum Development, *Curriculum Guides, *Drug Abuse, Elementary School Students, Instructional Materials, Secondary School Students, *Social Problems, *Teaching Guides ABSTRACT This very extensive guide, designed in large measure by classroom teachers an' meant for use by classroom teachers, is one community's response to its drug problem. The completed guide, however, is designed for adaptation throughout the nation and in foreign countries. Material is offered for different school levels, with the primary grades receiving information introduced by the classroom teacher, focusing on mental health and character development. The concept of drugs as medicine and narcotics is presented at the upper elementary level. The approach in the secondary grades is through the established curriculum with units offered in English, Mathematics, Science, Health and Physical Education and Social Studies. Specific yet flexible guidelines are included at each grade level to help establish objectives, create motivation, and provide activities for enrichment and reinforcement. Glossaries and factual information which can help answer questions often asked are included, as well as letters to committee members and parents. (CJ) Laredo Independent School District C\I C\J CD W THE USE, MISUSE, AND ABUSE OF DRUGS AND NARCOTICS SECOND EDITION r-4 CV CV O Final Progress Report Proposal No. -

(EUA) of VEKLURY® (Remdesivir) for HOSPITALIZED PEDIATRIC
FACT SHEET FOR HEALTHCARE PROVIDERS EMERGENCY USE AUTHORIZATION (EUA) OF VEKLURY® (remdesivir) FOR HOSPITALIZED PEDIATRIC PATIENTS WEIGHING 3.5 KG TO LESS THAN 40 KG OR HOSPITALIZED PEDIATRIC PATIENTS LESS THAN 12 YEARS OF AGE WEIGHING AT LEAST 3.5 KG The U.S. Food and Drug Administration (FDA) has issued an Emergency Use Authorization (EUA) to permit the emergency use of VEKLURY for treatment of suspected or laboratory confirmed coronavirus disease 2019 (COVID-19) in hospitalized pediatric patients weighing 3.5 kg to less than 40 kg or hospitalized pediatric patients less than 12 years of age weighing at least 3.5 kg. VEKLURY has been authorized by FDA for the emergency uses described above. VEKLURY is not FDA-approved for these uses. VEKLURY is authorized only for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of VEKLURY under section 564(b)(1) of the Act, 21 U.S.C. § 360bbb-3(b)(1), unless the authorization is terminated or revoked sooner. This EUA is for the use of VEKLURY to treat COVID-19 in hospitalized pediatric patients weighing 3.5 kg to less than 40 kg or hospitalized pediatric patients less than 12 years of age weighing at least 3.5 kg. VEKLURY must be administered by intravenous (IV) infusion. Healthcare providers must submit a report on all medication errors and ALL SERIOUS ADVERSE EVENTS related to VEKLURY. See Sections 8 and 9 of the Full EUA Prescribing Information for reporting requirements. • See the Full EUA Prescribing Information for complete dosage, preparation, and administration instructions. -

Hospital Standard File Pricing Transparency 2020 0.Pdf
Phoenix Children's Hospital Hospital Standard File Pricing as of Jan. 1, 2020 Service Code Description Rev Code Price 37010001 GEN SURG LEV 1 1ST HR 360 4,222.00 37010002 GEN SURG LEV 1 ADD 1/2 HR 360 2,112.00 37010005 GEN SURG LEV 2 1ST HR 360 6,547.00 37010007 GEN SURG LEV 2 ADD 1/2 HR 360 3,273.00 37010009 GEN SURG LEV 3 1ST HR 360 8,325.00 37010010 GEN SURG LEV 3 ADD 1/2 HR 360 4,163.00 37010011 BAG AUTOTRANS ATRIUM 272 263.00 37010013 GEN SURG LEV 4 1ST HR 360 11,747.00 37010015 GEN SURG LEV 4 ADD 1/2 HR 360 5,874.00 37010019 GEN SURG LEV 5 ADD 1/2 HR 360 7,036.00 37010020 GEN SURG LEV 5 1ST HR 360 14,072.00 37010021 LITHOTRIPSY 1ST HR 790 7,672.00 37010023 LITHOTRIPSY ADD 1/2 HR 790 3,837.00 37010101 BLOOD TRANSFUSN 391 1,763.00 37010105 IV INF HYDRAT ADDL HR 260 305.00 37010107 IV INF EA ADDL HR 260 305.00 37010108 IV INF EA ADDL DRUG 260 339.00 37010116 CHEMO ADMIN IV PUSH INIT 331 687.00 37010117 CHEMO ADMIN IV PUSH ADD 331 418.00 37010118 CHEMO INF 1ST HR 335 1,071.00 37010119 CHEMO INF EA ADDL HR 335 415.00 37010121 CHEMO INF EA ADDL DRUG 335 631.00 37010231 US GUIDE NDL PLCMT S&I 402 1,008.00 37010233 US GUIDE VAD S&I 402 929.00 37010234 REP RECRRNT INCISI HERNIA 360 25,888.00 37010264 N BLCK INJ BRACHIAL PLEX 361 1,269.00 37010268 N BLCK INJ AXILLARY 361 1,269.00 37010270 N BLCK INJ SUPRASCAPULAR 361 1,269.00 37010282 N BLCK INJ SCIATIC SNG 361 1,269.00 37010284 N BLK INJ SCIATIC CONT IN 361 1,269.00 37010286 N BLCK INJ FEM SNGL 361 1,269.00 37010288 N BLCK INJ FEM CONT INF 361 1,269.00 37010292 N BLCK OTHER PERIPHERAL 361 -

Forensic Pharmacology__Inside
INSIDE FORENSIC SCIENCE Forensic Pharmacology INSIDE FORENSIC SCIENCE Forensic Anthropology Forensic DNA Analysis Forensic Medicine Forensic Pharmacology Legal Aspects of Forensics The Forensic Aspects of Poisons INSIDE FORENSIC SCIENCE Forensic Pharmacology Beth E. Zedeck, MSW, RN, MSN and Morris S. Zedeck, Ph.D. SERIES EDITOR | Lawrence Kobilinsky, Ph.D. The authors are proud to have worked as a father and daughter team on this project and wish to thank Dr. Zedeck’s wife, Ellen Lieberman, Esq., for her assistance and thoughtful suggestions during the preparation of this book. Forensic Pharmacology Copyright © 2007 by Infobase Publishing All rights reserved. No part of this book may be reproduced or utilized in any form or by any means, electronic or mechanical, including photocopying, recording, or by any information storage or retrieval systems, without permission in writing from the publisher. For information contact: Chelsea House An imprint of Infobase Publishing 132 West 31st Street New York NY 10001 Library of Congress Cataloging-in-Publication Data Zedeck, Beth E. Forensic pharmacology / Beth E. Zedeck and Morris S. Zedeck. p. cm. — (Inside forensic science) Includes bibliographical references and index. ISBN 0-7910-8920-7 (hardcover) 1. Forensic pharmacology. I. Zedeck, Morris S. II. Title. RA1160.Z43 2006 614'.1—dc22 2006020624 Chelsea House books are available at special discounts when purchased in bulk quantities for businesses, associations, institutions, or sales promotions. Please call our Special Sales Department in New York at (212) 967-8800 or (800) 322-8755. You can find Chelsea House on the World Wide Web at http://www.chelseahouse.com Cover design by Ben Peterson Text design by Annie O’Donnell Printed in the United States of America BANG FOF 10 9 8 7 6 5 4 3 2 1 This book is printed on acid-free paper. -

Prescribing Information for RISPERDAL CONSTA
RISPERDAL CONSTA® RISPERDAL CONSTA® (risperidone) LONG-ACTING INJECTION (risperidone) LONG-ACTING INJECTION • Hyperprolactinemia: Risperidone treatment may elevate prolactin levels. Long- HIGHLIGHTS OF PRESCRIBING INFORMATION standing hyperprolactinemia, when associated with hypogonadism, can lead These highlights do not include all the information needed to use to decreased bone density in men and women. (5.6) RISPERDAL CONSTA® safely and effectively. See full prescribing information • Orthostatic hypotension: associated with dizziness, tachycardia, bradycardia, for RISPERDAL CONSTA®. and syncope can occur, especially during initial dose titration with oral RISPERDAL CONSTA® (risperidone) LONG-ACTING INJECTION risperidone. Use caution in patients with cardiovascular disease, cerebrovascular disease, and conditions that could affect hemodynamic Initial U.S. Approval: 2003 responses. (5.7) • Leukopenia, Neutropenia, and Agranulocytosis have been reported with WARNING: INCREASED MORTALITY IN ELDERLY PATIENTS antipsychotics, including RISPERDAL CONSTA®. Patients with history of a WITH DEMENTIA-RELATED PSYCHOSIS clinically significant low white blood cell count (WBC) or a drug-induced See full prescribing information for complete boxed warning. leukopenia/neutropenia should have their complete blood cell count (CBC) Elderly patients with dementia-related psychosis treated with antipsychotic monitored frequently during the first few months of therapy and drugs are at an increased risk of death. RISPERDAL CONSTA® is not approved discontinuation of RISPERDAL CONSTA® should be considered at the first sign for use in patients with dementia-related psychosis. (5.1) of a clinically significant decline in WBC in the absence of other causative factors. (5.9) • Potential for cognitive and motor impairment: has potential to impair judgment, ---------------------------------RECENT MAJOR CHANGES-------------------------------- thinking, and motor skills. Use caution when operating machinery, including Warnings and Precautions (5.3, 5.4) 2/2021 automobiles. -

RISPERDAL CONSTA® Risperidone NEW ZEALAND DATA SHEET
RISPERDAL CONSTA® risperidone NEW ZEALAND DATA SHEET 1. PRODUCT NAME RISPERDAL CONSTA 25 mg/2 ml Suspension for injection RISPERDAL CONSTA 37.5 mg/2 ml Suspension for injection RISPERDAL CONSTA 50 mg/2 ml Suspension for injection 2. QUALITATIVE AND QUANTITATIVE COMPOSITION RISPERDAL CONSTA is an extended release microspheres formulation of risperidone micro- encapsulated in polyglactin for intramuscular injection, in strengths of 25mg, 37.5mg and 50mg when suspended in 2mL diluent. For a full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM Powder and solvent for prolonged-release suspension for injection. RISPERDAL CONSTA contains either 25 mg, 37.5 mg or 50 mg risperidone and is presented as a white to off-white free-flowing powder in a 5mL vial and a prefilled syringe containing 2mL diluent, together with: - One Vial Adapter for reconstitution (referred as Vial Adapter), and - Two Terumo SurGuard needles for intramuscular injection (a 21G UTW 1-inch safety needle with needle protection device for deltoid administration and a 20G TW 2-inch safety needle with needle protection device for gluteal administration). 4. CLINICAL PARTICULARS 4.1 Therapeutic indications RISPERDAL CONSTA is indicated for the treatment of schizophrenia and other psychotic disorders. These include first episode psychoses, acute schizophrenic exacerbations, chronic schizophrenia and other psychotic conditions, in which positive symptoms (such as hallucinations, delusions, thought disturbances, hostility, suspiciousness), and/or negative symptoms (such as blunted effect, emotional and social withdrawal, poverty of speech) are prominent. RISPERDAL CONSTA also alleviates affective symptoms (such as depression, guilt-feelings, anxiety) associated with schizophrenia. In addition, RISPERDAL CONSTA also appears effective in maintaining the clinical improvement during continuation therapy in patients who have shown an initial response to treatment with this agent.