The Importance of Study Design Abundant Features in High
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Human Catechol O-Methyltransferase Genetic Variation
Molecular Psychiatry (2004) 9, 151–160 & 2004 Nature Publishing Group All rights reserved 1359-4184/04 $25.00 www.nature.com/mp ORIGINAL RESEARCH ARTICLE Human catechol O-methyltransferase genetic variation: gene resequencing and functional characterization of variant allozymes AJ Shield1, BA Thomae1, BW Eckloff2, ED Wieben2 and RM Weinshilboum1 1Department of Molecular Pharmacology and Experimental Therapeutics, Mayo Medical School, Mayo Clinic, Mayo Foundation, Rochester, MN, USA; 2Department of Biochemistry and Molecular Biology, Mayo Medical School, Mayo Clinic, Mayo Foundation, Rochester, MN, USA Catechol O-methyltransferase (COMT) plays an important role in the metabolism of catecholamines, catecholestrogens and catechol drugs. A common COMT G472A genetic polymorphism (Val108/158Met) that was identified previously is associated with decreased levels of enzyme activity and has been implicated as a possible risk factor for neuropsychiatric disease. We set out to ‘resequence’ the human COMT gene using DNA samples from 60 African-American and 60 Caucasian-American subjects. A total of 23 single nucleotide polymorphisms (SNPs), including a novel nonsynonymous cSNP present only in DNA from African-American subjects, and one insertion/deletion were observed. The wild type (WT) and two variant allozymes, Thr52 and Met108, were transiently expressed in COS-1 and HEK293 cells. There was no significant change in level of COMT activity for the Thr52 variant allozyme, but there was a 40% decrease in the level of activity in cells transfected with the Met108 construct. Apparent Km values of the WT and variant allozymes for the two reaction cosubstrates differed slightly, but significantly, for 3,4-dihydroxybenzoic acid but not for S-adenosyl-L-methionine. -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota. -

EZH2 in Normal Hematopoiesis and Hematological Malignancies
www.impactjournals.com/oncotarget/ Oncotarget, Vol. 7, No. 3 EZH2 in normal hematopoiesis and hematological malignancies Laurie Herviou2, Giacomo Cavalli2, Guillaume Cartron3,4, Bernard Klein1,2,3 and Jérôme Moreaux1,2,3 1 Department of Biological Hematology, CHU Montpellier, Montpellier, France 2 Institute of Human Genetics, CNRS UPR1142, Montpellier, France 3 University of Montpellier 1, UFR de Médecine, Montpellier, France 4 Department of Clinical Hematology, CHU Montpellier, Montpellier, France Correspondence to: Jérôme Moreaux, email: [email protected] Keywords: hematological malignancies, EZH2, Polycomb complex, therapeutic target Received: August 07, 2015 Accepted: October 14, 2015 Published: October 20, 2015 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Enhancer of zeste homolog 2 (EZH2), the catalytic subunit of the Polycomb repressive complex 2, inhibits gene expression through methylation on lysine 27 of histone H3. EZH2 regulates normal hematopoietic stem cell self-renewal and differentiation. EZH2 also controls normal B cell differentiation. EZH2 deregulation has been described in many cancer types including hematological malignancies. Specific small molecules have been recently developed to exploit the oncogenic addiction of tumor cells to EZH2. Their therapeutic potential is currently under evaluation. This review summarizes the roles of EZH2 in normal and pathologic hematological processes and recent advances in the development of EZH2 inhibitors for the personalized treatment of patients with hematological malignancies. PHYSIOLOGICAL FUNCTIONS OF EZH2 state through tri-methylation of lysine 27 on histone H3 (H3K27me3) [6]. -
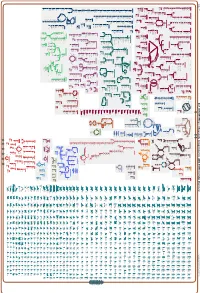
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Ron Caspi Peter Midford Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_003855395Cyc: Shewanella livingstonensis LMG 19866 Cellular Overview Connections between pathways are omitted for legibility. -

Expanding the Structural Diversity of DNA Methyltransferase Inhibitors
pharmaceuticals Article Expanding the Structural Diversity of DNA Methyltransferase Inhibitors K. Eurídice Juárez-Mercado 1 , Fernando D. Prieto-Martínez 1 , Norberto Sánchez-Cruz 1 , Andrea Peña-Castillo 1, Diego Prada-Gracia 2 and José L. Medina-Franco 1,* 1 DIFACQUIM Research Group, Department of Pharmacy, School of Chemistry, National Autonomous University of Mexico, Avenida Universidad 3000, Mexico City 04510, Mexico; [email protected] (K.E.J.-M.); [email protected] (F.D.P.-M.); [email protected] (N.S.-C.); [email protected] (A.P.-C.) 2 Research Unit on Computational Biology and Drug Design, Children’s Hospital of Mexico Federico Gomez, Mexico City 06720, Mexico; [email protected] * Correspondence: [email protected] Abstract: Inhibitors of DNA methyltransferases (DNMTs) are attractive compounds for epigenetic drug discovery. They are also chemical tools to understand the biochemistry of epigenetic processes. Herein, we report five distinct inhibitors of DNMT1 characterized in enzymatic inhibition assays that did not show activity with DNMT3B. It was concluded that the dietary component theaflavin is an inhibitor of DNMT1. Two additional novel inhibitors of DNMT1 are the approved drugs glyburide and panobinostat. The DNMT1 enzymatic inhibitory activity of panobinostat, a known pan inhibitor of histone deacetylases, agrees with experimental reports of its ability to reduce DNMT1 activity in liver cancer cell lines. Molecular docking of the active compounds with DNMT1, and re-scoring with the recently developed extended connectivity interaction features approach, led to an excellent agreement between the experimental IC50 values and docking scores. Citation: Juárez-Mercado, K.E.; Keywords: dietary component; epigenetics; enzyme inhibition; focused library; epi-informatics; Prieto-Martínez, F.D.; Sánchez-Cruz, multitarget epigenetic agent; natural products; chemoinformatics N.; Peña-Castillo, A.; Prada-Gracia, D.; Medina-Franco, J.L. -

Catechol-O-Methyltransferase Inhibitor, on the Motor Response to Acute Treatment with Levodopa in Patients with Parkinson's Disease
18616ournal ofNeurology, Neurosurgery, and Psychiatry 1994;57:186-189 J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.57.2.186 on 1 February 1994. Downloaded from Effect of entacapone, a peripherally acting catechol-O-methyltransferase inhibitor, on the motor response to acute treatment with levodopa in patients with Parkinson's disease Marcelo Merello, Andrew J Lees, Roy Webster, Michael Bovingdon, Ariel Gordin Abstract treatment.7-'0 The oral bioavailability of lev- Catechol-O-methyltransferase (COMT) odopa is low due to first pass metabolism inhibitors may be useful in the treatment which is at least partly due to the high activity of Parkinson's disease by improving the of COMT in the gut and liver." 12 The com- bioavailability of levodopa and by pro- bination of a peripheral COMT inhibitor longing its effects. Entacapone (OR-611), with levodopa/DCI therapy might therefore a novel COMT inhibitor, which does not produce a smoother and more prolonged cross the blood brain barrier, was motor response in patients with Parkinson's assessed in 12 patients with Parkinson's disease."3 14 As early as in 1971 Ericsson'5 disease and motor fluctuations in a reported beneficial effects with a COMT randomised, double-blind, cross-over, inhibitor with concomitant reduction in single dose study. The magnitude and levodopa-induced dyskinesias. More recently, duration of the therapeutic response to a a new generation of COMT inhibitors have single dose of 200 mg levodopa/50 mg been developed, which are much more potent carbidopa was evaluated after concomi- and specific."'3 1620 Fluorodopa positron tant placebo, or 200 or 800 mg enta- emission tomography (PET) studies in mon- capone. -

Mechanism of N-Methylation by the Trna M1g37 Methyltransferase Trm5
Thomas Jefferson University Jefferson Digital Commons Department of Biochemistry and Molecular Department of Biochemistry and Molecular Biology Faculty Papers Biology 12-1-2010 Mechanism of N-methylation by the tRNA m1G37 methyltransferase Trm5. Thomas Christian Thomas Jefferson University Georges Lahoud Thomas Jefferson University Cuiping Liu Thomas Jefferson University Katherine Hoffmann University of California at Santa Barbara Follow this and additional works at: https://jdc.jefferson.edu/bmpfp John J Perona Univ Persityart of of the California Biochemistr at Santay Commons Barbara, Medical Biochemistry Commons, and the Molecular Biology Commons SeeLet next us page know for additional how authors access to this document benefits ouy Recommended Citation Christian, Thomas; Lahoud, Georges; Liu, Cuiping; Hoffmann, Katherine; Perona, John J; and Hou, Ya-Ming, "Mechanism of N-methylation by the tRNA m1G37 methyltransferase Trm5." (2010). Department of Biochemistry and Molecular Biology Faculty Papers. Paper 26. https://jdc.jefferson.edu/bmpfp/26 This Article is brought to you for free and open access by the Jefferson Digital Commons. The Jefferson Digital Commons is a service of Thomas Jefferson University's Center for Teaching and Learning (CTL). The Commons is a showcase for Jefferson books and journals, peer-reviewed scholarly publications, unique historical collections from the University archives, and teaching tools. The Jefferson Digital Commons allows researchers and interested readers anywhere in the world to learn about and keep up to date with Jefferson scholarship. This article has been accepted for inclusion in Department of Biochemistry and Molecular Biology Faculty Papers by an authorized administrator of the Jefferson Digital Commons. For more information, please contact: [email protected]. -

Arylamine N-Methyltransferase and Thiol Methyltransferase Activities in Cholestatic Rat Liver Induced by Common Bile Duct Ligation
EXPERIMENTAL and MOLECULAR MEDICINE, Vol. 33, No. 1, 23-28, March 2001 Arylamine N-methyltransferase and thiol methyltransferase activities in cholestatic rat liver induced by common bile duct ligation You Hee Kim1,2 and Il Joo1 using tryptamine or 4-chlorothiophenol as substrates and S-Adenosyl-L-[methyl-3H]methionine as co-sub- 1 Department of Biochemistry, Keimyung University School of strate. The results indicate that although the Km val- Medicine, Taegu 700-712, Korea ues were about the same as the sham-operated 2 Corresponding author: Tel, +82-53-250-7462; control, the Vmax values of both enzymes increased Fax, +82-53-252-1605; E-mail, [email protected] significantly (Pú0.01 and 0.001, respectively). These results suggest that the biosynthesis of arylamine Accepted 2 March, 2001 N-methyltransferase and thiol methyltransferase have been induced in response to obstructive jaun- dice. Abstract Keywords: arylamine N-methyltransferase, cholestatic Methylation catalyzed by methyltransferases is a rat liver, enzyme induction, thiol methyltransferase major metabolic pathway for an inactivation of some catecholamines, niacinamide as well as aliphatic sulfhydryl drugs and toxic hydrogen sulfides. To investigate the effects of obstructive jaundice in an Introduction animal model, common bile duct ligation (CBDL) was performed in the rat and enzyme activities of S- Methylation is a major metabolic pathway for an inacti- adenosyl-L-methionine-dependent arylamine N- vation of some catecholamines, niacinamide as well as methyltransferase and thiol methyltransferase were aliphatic sulfhydryl drugs and toxic hydrogen sulfides examined in liver cell fractions and serum for a catalyzed by methyltransferases (N-methyltransferases period of 42 d after CBDL. -

5-Methylcytidine Has a Complex, Context-Dependent Role in RNA
5-methylcytidine has a complex, context-dependent role in RNA Andrew Mark Shafik April 2017 A thesis submitted for the degree of Doctor of Philosophy The Australian National University Originality Statement I hereby declare that this submission is my own work and to the best of my knowledge it contains no materials written by another person, or substantial proportions of material which have been accepted for the award of any other degree or diploma at ANU or any other educational institution, except where due acknowledgement is made in this thesis. Any contribution made to the research by others, with whom I have worked at ANU or elsewhere, is explicitly acknowledged in this thesis. I also declare that the intellectual content of this thesis is the product of my own work, except to the extent that assistance from others in the project’s design and conception or in style, presentation and linguistic expression is acknowledged. Signed ………………………. Date …………………………. II Acknowledgements I am overwhelmed. I have finally completed this milestone, three and a half years of my life culminating in this body of work. The path hasn’t been easy but I have finally reached the end. Of course, this would not have been possible without the support of many people. To my supervisor, Thomas Preiss, thank you for reading the thesis and the stimulating discussions. To Maurits Evers, thank you so much for the collaboration, for the bioinformatics support concerning the metagene analyses. This gave another dimension to the thesis. Thanks for reading my thesis, for the many valuable discussion we had, and for the coffee breaks. -

Histone Methyltransferase Activity/Inhibition
www.abnova.com Introduction and Background A. Overview Epigenetic inactivation of genes plays a critical role in many important human diseases, especially in cancer. A major mechanism for epigenetic inactivation of the genes is methylation of CpG islands in genome DNA caused by DNA methyltransferases.Histone methyltransferases (HMTs) control or regulate DNA methylation through chromatin-dependent transcription repression or activation. HMTs transfer 1-3 methyl groups from S-adenosyl-L-methionine to the lysine and arginine residues of histone proteins. Inhibition of HMTs may lead to expression of the silenced genes and HMT inhibitors are currently developed for various therapeutic or experimental applications. ESET, G9a, SUV39-h1, SUV39-h2, SETDB1, Dim-5 and Eu-HMTase are histone methyltransferases that catalyze methylation of histone H3 at lysine 9 (H3-K9) in mammalian cells. H3-K9 methylation mediates heterochromatin formation by forming a binding site for HP1 and also participates in silencing gene expression at euchromatic sites. There is only the radioisotopic method currently available for measuring HMT activity/inhibition, which is time consuming, labor-intensive, and has low throughput or produces radioactive waste. The Histone Methyltransferase Activity/Inhibition Assay Kit (H3-K9) addresses these problems by using a unique procedure to measure HMT activity/inhibition. The kit has the following features: Quick and efficient, which can be finished within 3 hours. Innovative colorimetric assay with no need for radioactivity, electrophoresis, and chromatography. Specific measurement of activity/inhibition of H3-K9 histone methyltransferases. Strip microplate format makes the assay flexible: manual or high throughput analysis. Simple, reliable, and consistent assay conditions. B. -

A Second DNA Methyltransferase Repair Enzyme in Escherichia Coli (Ada-Alb Operon Deletion/O'-Methylguanine/04-Methylthyne/Suicide Enzyme) G
Proc. Nati. Acad. Sci. USA Vol. 85, pp. 3039-3043, May 1988 Genetics A second DNA methyltransferase repair enzyme in Escherichia coli (ada-alB operon deletion/O'-methylguanine/04-methylthyne/suicide enzyme) G. WILLIAM REBECK, SUSAN COONS, PATRICK CARROLL, AND LEONA SAMSON* Charles A. Dana Laboratory of Toxicology, Harvard School of Public Health, Boston, MA 02115 Communicated by Elkan R. Blout, December 28, 1987 (receivedfor review November 9, 1987) ABSTRACT The Escherichia coli ada-akB operon en- ada gene encodes a 39-kDa DNA methyltransferase with two codes a 39-kDa protein (Ada) that is a DNA-repair methyl- active sites, one that removes methyl groups from O6- transferase and a 27-kDa protein (AlkB) of unknown function. methylguanine (06-MeGua) or 04-methylthymine (04- By DNA blot hybridization analysis we show that the alkyla- MeThy) and one that removes methyl groups from methyl tion-sensitive E. cofi mutant BS23 [Sedgwick, B. & Lindahl, T. phosphotriester lesions (7, 12-15). The Ada protein is one of (1982) J. Mol. Biol. 154, 169-1751 is a deletion mutant lacking several gene products to be induced as E. coli adapt to the entire ada-aik operon. Despite the absence ofthe ada gene become alkylation-resistant upon exposure to low doses of and its product, the cells contain detectable levels of a DNA- alkylating agents (3, 12, 13). The repair of methyl phospho- repair methyltransferase activity. We conclude that the meth- triester lesions converts the Ada protein into a positive yltransferase in BS23 cells is the product of a gene other than regulator ofthe ada gene, and this adaptive response (16) and ada. -

COMT) Haplotypes with Nicotine Dependence in Male and Female Smokers of Two Ethnic Populations
Neuropsychopharmacology (2006) 31, 675–684 & 2006 Nature Publishing Group All rights reserved 0893-133X/06 $30.00 www.neuropsychopharmacology.org Significant Association of Catechol-O-Methyltransferase (COMT) Haplotypes with Nicotine Dependence in Male and Female Smokers of Two Ethnic Populations Joke Beuten1, Thomas J Payne2, Jennie Z Ma1 and Ming D Li*,1 1 Program in Genomics and Bioinformatics on Drug Addiction, Department of Psychiatry, The University of Texas Health Science Center at San Antonio, San Antonio, TX, USA; 2The ACT Tobacco Center, The University of Mississippi School of Dentistry, Jackson, MS, USA The catechol-O-methyltransferase (COMT) gene plays a prominent role in dopaminergic circuits central to drug reward. Allelic variants within the COMT gene are therefore potential candidates for examining interindividual differences in vulnerability to nicotine dependence (ND). We analyzed five single nucleotide polymorphisms (SNPs), including the Val/Met variant (rs4680), which results in a three- to four- fold difference in enzyme activity within COMT, for association with the three ND measures, SQ, HSI, and FTND, in 602 nuclear families of African-American (AA) or European-American (EA) origin. The Val/Met variant showed a significant association with the three ND measures in the pooled and EA samples and with FTND in the AA sample. Haplotype analysis revealed a major protective A-G-T haplotype (frequency 23.6%) for rs740603-rs4680-rs174699 in the AA sample (minimum Z ¼À3.35; P ¼ 0.0005 for FTND), a major protective T-G-T haplotype (frequency 15.2%; minimum Z ¼À2.92; P ¼ 0.003 for FTND) in the EA sample, and a high-risk C-A-T haplotype (frequency 16.9%; minimum Z ¼ 3.16; P ¼ 0.002 for SQ) in the AA sample for rs933271-rs4680-rs174699.