Expanding the Structural Diversity of DNA Methyltransferase Inhibitors
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Structural and Functional Coordination of DNA and Histone Methylation
Structural and Functional Coordination of DNA and Histone Methylation Xiaodong Cheng Department of Biochemistry, Emory University School of Medicine, Atlanta, Georgia 30322 Correspondence: [email protected] SUMMARY One of the most fundamental questions in the control of gene expression in mammals is how epigenetic methylation patterns of DNA and histones are established, erased, and recognized. This central process in controlling gene expression includes coordinated covalent modifications of DNA and its associated histones. This article focuses on structural aspects of enzymatic activities of histone (arginine and lysine) methylation and demethylation and functional links between the methylation status of the DNA and histones. An interconnected network of methyltransferases, demethylases, and accessory proteins is responsible for changing or maintaining the modification status of specific regions of chromatin. Outline 1 Histone methylation and demethylation 4 Summary 2 DNA methylation References 3 Interplay between DNA methylation and histone modification Editors: C. David Allis, Marie-Laure Caparros, Thomas Jenuwein, and Danny Reinberg Additional Perspectives on Epigenetics available at www.cshperspectives.org Copyright # 2014 Cold Spring Harbor Laboratory Press; all rights reserved; doi: 10.1101/cshperspect.a018747 Cite this article as Cold Spring Harb Perspect Biol 2014;6:a018747 1 X. Cheng OVERVIEW All cells face the problem of controlling the amounts and articles by Becker and Workman 2013; Allis et al. 2014; timing of expression of their -

Human Catechol O-Methyltransferase Genetic Variation
Molecular Psychiatry (2004) 9, 151–160 & 2004 Nature Publishing Group All rights reserved 1359-4184/04 $25.00 www.nature.com/mp ORIGINAL RESEARCH ARTICLE Human catechol O-methyltransferase genetic variation: gene resequencing and functional characterization of variant allozymes AJ Shield1, BA Thomae1, BW Eckloff2, ED Wieben2 and RM Weinshilboum1 1Department of Molecular Pharmacology and Experimental Therapeutics, Mayo Medical School, Mayo Clinic, Mayo Foundation, Rochester, MN, USA; 2Department of Biochemistry and Molecular Biology, Mayo Medical School, Mayo Clinic, Mayo Foundation, Rochester, MN, USA Catechol O-methyltransferase (COMT) plays an important role in the metabolism of catecholamines, catecholestrogens and catechol drugs. A common COMT G472A genetic polymorphism (Val108/158Met) that was identified previously is associated with decreased levels of enzyme activity and has been implicated as a possible risk factor for neuropsychiatric disease. We set out to ‘resequence’ the human COMT gene using DNA samples from 60 African-American and 60 Caucasian-American subjects. A total of 23 single nucleotide polymorphisms (SNPs), including a novel nonsynonymous cSNP present only in DNA from African-American subjects, and one insertion/deletion were observed. The wild type (WT) and two variant allozymes, Thr52 and Met108, were transiently expressed in COS-1 and HEK293 cells. There was no significant change in level of COMT activity for the Thr52 variant allozyme, but there was a 40% decrease in the level of activity in cells transfected with the Met108 construct. Apparent Km values of the WT and variant allozymes for the two reaction cosubstrates differed slightly, but significantly, for 3,4-dihydroxybenzoic acid but not for S-adenosyl-L-methionine. -

From 1957 to Nowadays: a Brief History of Epigenetics
International Journal of Molecular Sciences Review From 1957 to Nowadays: A Brief History of Epigenetics Paul Peixoto 1,2, Pierre-François Cartron 3,4,5,6,7,8, Aurélien A. Serandour 3,4,6,7,8 and Eric Hervouet 1,2,9,* 1 Univ. Bourgogne Franche-Comté, INSERM, EFS BFC, UMR1098, Interactions Hôte-Greffon-Tumeur/Ingénierie Cellulaire et Génique, F-25000 Besançon, France; [email protected] 2 EPIGENEXP Platform, Univ. Bourgogne Franche-Comté, F-25000 Besançon, France 3 CRCINA, INSERM, Université de Nantes, 44000 Nantes, France; [email protected] (P.-F.C.); [email protected] (A.A.S.) 4 Equipe Apoptose et Progression Tumorale, LaBCT, Institut de Cancérologie de l’Ouest, 44805 Saint Herblain, France 5 Cancéropole Grand-Ouest, Réseau Niches et Epigénétique des Tumeurs (NET), 44000 Nantes, France 6 EpiSAVMEN Network (Région Pays de la Loire), 44000 Nantes, France 7 LabEX IGO, Université de Nantes, 44000 Nantes, France 8 Ecole Centrale Nantes, 44300 Nantes, France 9 DImaCell Platform, Univ. Bourgogne Franche-Comté, F-25000 Besançon, France * Correspondence: [email protected] Received: 9 September 2020; Accepted: 13 October 2020; Published: 14 October 2020 Abstract: Due to the spectacular number of studies focusing on epigenetics in the last few decades, and particularly for the last few years, the availability of a chronology of epigenetics appears essential. Indeed, our review places epigenetic events and the identification of the main epigenetic writers, readers and erasers on a historic scale. This review helps to understand the increasing knowledge in molecular and cellular biology, the development of new biochemical techniques and advances in epigenetics and, more importantly, the roles played by epigenetics in many physiological and pathological situations. -

The Role of B-Vitamins – Gene Interactions in Colorectal Carcinogenesis a Molecular Epidemiological Approach
THE ROLE OF B-VITAMINS – GENE INTERACTIONS IN COLORECTAL CARCINOGENESIS A MOLECULAR EPIDEMIOLOGICAL APPROACH Promotor Prof. dr. ir. F.J. Kok Hoogleraar Voeding en Gezondheid Wageningen Universiteit Co-promotoren Dr. ir. E. Kampman Universitair Hoofddocent, sectie Humane Voeding Wageningen Universiteit Dr. J. Keijer Hoofd Food Bioactives Group RIKILT – Instituut voor Voedselveiligheid Promotiecommissie Prof. dr. J. Mathers University of Newcastle Upon Tyne, United Kingdom Prof. dr. G.A. Meijer VU Medisch Centrum, Amsterdam Dr. ir. P. Verhoef Wageningen Centre for Food Sciences Prof. dr. S.C. de Vries Wageningen Universiteit Dit onderzoek is uitgevoerd binnen de onderzoeksschool VLAG. THE ROLE OF B-VITAMINS – GENE INTERACTIONS IN COLORECTAL CARCINOGENESIS A MOLECULAR EPIDEMIOLOGICAL APPROACH Maureen van den Donk Proefschrift ter verkrijging van de graad van doctor op gezag van de rector magnificus van Wageningen Universiteit, Prof. dr. M.J. Kropff, in het openbaar te verdedigen op dinsdag 13 december 2005 des namiddags te half twee in de Aula Maureen van den Donk The role of B-vitamins – gene interactions in colorectal carcinogenesis – A molecular epidemiological approach Thesis Wageningen University – With references – With summary in Dutch ISBN 90-8504-328-X ABSTRACT The role of B-vitamins–gene interactions in colorectal carcinogenesis – A molecular epidemiological approach PhD thesis by Maureen van den Donk, Division of Human Nutrition, Wageningen University, The Netherlands, December 13, 2005. Folate deficiency can affect DNA methylation and DNA synthesis. Both factors may be operative in colorectal carcinogenesis. Many enzymes, like methylenetetrahydrofolate reductase (MTHFR), thymidylate synthase (TS), methionine synthase (MTR), and serine hydroxymethyltransferase (SHMT), are needed for conversions in folate metabolism. -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota. -

Stress-Related Methylation of the Catechol-O-Methyltransferase Val158 Allele Predicts Human Prefrontal Cognition and Activity
6692 • The Journal of Neuroscience, May 4, 2011 • 31(18):6692–6698 Behavioral/Systems/Cognitive Stress-Related Methylation of the Catechol-O-Methyltransferase Val158 Allele Predicts Human Prefrontal Cognition and Activity Gianluca Ursini,1,2 Valentina Bollati,3,4 Leonardo Fazio,1 Annamaria Porcelli,1 Luisa Iacovelli,5 Assia Catalani,5 Lorenzo Sinibaldi,2 Barbara Gelao,1 Raffaella Romano,1 Antonio Rampino,1 Paolo Taurisano,1 Marina Mancini,1 Annabella Di Giorgio,1,6 Teresa Popolizio,6 Andrea Baccarelli,3,4,7 Antonio De Blasi,8 Giuseppe Blasi,1 and Alessandro Bertolino1,6 1Psychiatric Neuroscience Group, Department of Neurological and Psychiatric Sciences, University “Aldo Moro”, 71024 Bari, Italy, 2Mendel Laboratory, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) “Casa Sollievo della Sofferenza”, 71013 San Giovanni Rotondo, Italy, 3Center for Molecular and Genetic Epidemiology, Department of Environmental and Occupational Health, University of Milan, Milan, Italy, 4Istituto Di Ricovero e Cura a Carattere Scientifico Maggiore Hospital, Mangiagalli and Regina Elena Foundation, 20122 Milan, Italy, 5Department of Physiology and Pharmacology “V. Erspamer”, University of Rome “Sapienza”, 00185 Rome, Italy, 6Department of Neuroradiology, IRCCS “Casa Sollievo della Sofferenza”, 71013 San Giovanni Rotondo, Italy, 7Exposure, Epidemiology, and Risk Program, Department of Environmental Health, Harvard School of Public Health, Boston, Massachusetts 02115- 6018, and 8Department of Molecular Medicine, University of Rome “Sapienza”, 00185 Rome, Italy DNA methylation at CpG dinucleotides is associated with gene silencing, stress, and memory. The catechol-O-methyltransferase (COMT) Val 158 allele in rs4680 is associated with differential enzyme activity, stress responsivity, and prefrontal activity during working memory (WM), and it creates a CpG dinucleotide. -

EZH2 in Normal Hematopoiesis and Hematological Malignancies
www.impactjournals.com/oncotarget/ Oncotarget, Vol. 7, No. 3 EZH2 in normal hematopoiesis and hematological malignancies Laurie Herviou2, Giacomo Cavalli2, Guillaume Cartron3,4, Bernard Klein1,2,3 and Jérôme Moreaux1,2,3 1 Department of Biological Hematology, CHU Montpellier, Montpellier, France 2 Institute of Human Genetics, CNRS UPR1142, Montpellier, France 3 University of Montpellier 1, UFR de Médecine, Montpellier, France 4 Department of Clinical Hematology, CHU Montpellier, Montpellier, France Correspondence to: Jérôme Moreaux, email: [email protected] Keywords: hematological malignancies, EZH2, Polycomb complex, therapeutic target Received: August 07, 2015 Accepted: October 14, 2015 Published: October 20, 2015 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Enhancer of zeste homolog 2 (EZH2), the catalytic subunit of the Polycomb repressive complex 2, inhibits gene expression through methylation on lysine 27 of histone H3. EZH2 regulates normal hematopoietic stem cell self-renewal and differentiation. EZH2 also controls normal B cell differentiation. EZH2 deregulation has been described in many cancer types including hematological malignancies. Specific small molecules have been recently developed to exploit the oncogenic addiction of tumor cells to EZH2. Their therapeutic potential is currently under evaluation. This review summarizes the roles of EZH2 in normal and pathologic hematological processes and recent advances in the development of EZH2 inhibitors for the personalized treatment of patients with hematological malignancies. PHYSIOLOGICAL FUNCTIONS OF EZH2 state through tri-methylation of lysine 27 on histone H3 (H3K27me3) [6]. -
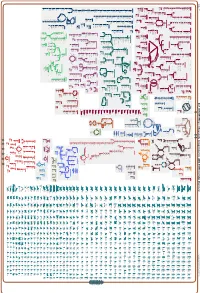
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Ron Caspi Peter Midford Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_003855395Cyc: Shewanella livingstonensis LMG 19866 Cellular Overview Connections between pathways are omitted for legibility. -
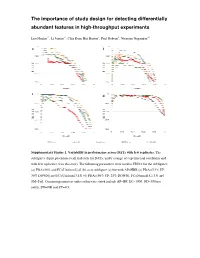
The Importance of Study Design Abundant Features in High
The importance of study design for detecting differentially abundant features in high -throughput experiments Luo Huaien 1* , Li Juntao 1*, C hia Kuan Hui Burton 1, Paul Robson 2, Niranjan Nagarajan 1# a b c d Supplementary Figure 1. Variability in performance across DATs with few repreplicates.licates. The subfigures depict precision-recall tradeoffs for DATs, under a range of experimexperimenentaltal conditions and with few replicates (2 in this case). The following parameters were used in EDDA for the sub figures: (a) PDA=30% and FC=Uniform[2,6] (b) as in subfigure (a) but with AP=HBR (c) PDA= [15% UP, 30% DOWN] and FC=Uniform[3,15] (d) PDA=[50% UP, 25% DOWN], FC=Normal[5,1.33] and SM=Full. Common parameters unless otherwise stated include AP=BP, EC=1000, ND=500 per entity, SM=NB and SV=0.5. a b c d Supplementary Figure 2 . Variability in performance across DATs with few data -points. Note that as in Supplementary Figure 1 , the performance of DATs varies widely merely by changing the number of replicates from 1 to 4 in sub figures (a)-(d) in a situation where the number data -points generated is limited (only 50 on average per entity). Results reported are based on data generated in EDDA with the following parameters (roughly mimicking a microbial RNA-seq application) : EC=1000, PDA=26%, FC=Uniform[3,7 ], AP=BP, SM=Full and SV=0.85. Supplementary Figure 3. Distribution of relative abundance in different empirically derived profiles. Note that both axes are plotted on a log-scale and that while BP does not have entities with relative abundance < 1e-5, the profile from Wu et al is more enriched for entities with relative abundance < 1e-5 than the HBR profile. -

Catechol-O-Methyltransferase Inhibitor, on the Motor Response to Acute Treatment with Levodopa in Patients with Parkinson's Disease
18616ournal ofNeurology, Neurosurgery, and Psychiatry 1994;57:186-189 J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.57.2.186 on 1 February 1994. Downloaded from Effect of entacapone, a peripherally acting catechol-O-methyltransferase inhibitor, on the motor response to acute treatment with levodopa in patients with Parkinson's disease Marcelo Merello, Andrew J Lees, Roy Webster, Michael Bovingdon, Ariel Gordin Abstract treatment.7-'0 The oral bioavailability of lev- Catechol-O-methyltransferase (COMT) odopa is low due to first pass metabolism inhibitors may be useful in the treatment which is at least partly due to the high activity of Parkinson's disease by improving the of COMT in the gut and liver." 12 The com- bioavailability of levodopa and by pro- bination of a peripheral COMT inhibitor longing its effects. Entacapone (OR-611), with levodopa/DCI therapy might therefore a novel COMT inhibitor, which does not produce a smoother and more prolonged cross the blood brain barrier, was motor response in patients with Parkinson's assessed in 12 patients with Parkinson's disease."3 14 As early as in 1971 Ericsson'5 disease and motor fluctuations in a reported beneficial effects with a COMT randomised, double-blind, cross-over, inhibitor with concomitant reduction in single dose study. The magnitude and levodopa-induced dyskinesias. More recently, duration of the therapeutic response to a a new generation of COMT inhibitors have single dose of 200 mg levodopa/50 mg been developed, which are much more potent carbidopa was evaluated after concomi- and specific."'3 1620 Fluorodopa positron tant placebo, or 200 or 800 mg enta- emission tomography (PET) studies in mon- capone. -

FULLTEXT01.Pdf
ARTICLE https://doi.org/10.1038/s41467-019-11179-9 OPEN Genome-wide systematic identification of methyltransferase recognition and modification patterns Torbjørn Ølshøj Jensen1, Christian Tellgren-Roth2, Stephanie Redl1,3, Jérôme Maury1, Simo Abdessamad Baallal Jacobsen1, Lasse Ebdrup Pedersen 1 & Alex Toftgaard Nielsen 1 1234567890():,; Genome-wide analysis of DNA methylation patterns using single molecule real-time DNA sequencing has boosted the number of publicly available methylomes. However, there is a lack of tools coupling methylation patterns and the corresponding methyltransferase genes. Here we demonstrate a high-throughput method for coupling methyltransferases with their respective motifs, using automated cloning and analysing the methyltransferases in vectors carrying a strain-specific cassette containing all potential target sites. To validate the method, we analyse the genomes of the thermophile Moorella thermoacetica and the mesophile Acetobacterium woodii, two acetogenic bacteria having substantially modified genomes with 12 methylation motifs and a total of 23 methyltransferase genes. Using our method, we characterize the 23 methyltransferases, assign motifs to the respective enzymes and verify activity for 11 of the 12 motifs. 1 The Novo Nordisk Foundation Center for Biosustainability (CfB), Technical University of Denmark (DTU), DK-2800 Lyngby, Denmark. 2 Uppsala Genome Center, National Genomics Infrastructure, SciLifeLab, SE-751 08 Uppsala, Sweden. 3 Massachusetts Institute of Technology, Cambridge, MA 02139, USA. Correspondence -

Histone Lysine Methyltransferases As Anti-Cancer Targets for Drug Discovery
Acta Pharmacologica Sinica (2016) 37: 1273–1280 © 2016 CPS and SIMM All rights reserved 1671-4083/16 www.nature.com/aps Review Histone lysine methyltransferases as anti-cancer targets for drug discovery Qing LIU1, 2, 3, Ming-wei WANG1, 2, 3, * 1The CAS Key Laboratory of Receptor Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China; 2The National Center for Drug Screening, Shanghai 201203, China; 3School of Pharmacy, Fudan University, Shanghai 201203, China Post-translational epigenetic modification of histones is controlled by a number of histone-modifying enzymes. Such modification regulates the accessibility of DNA and the subsequent expression or silencing of a gene. Human histone methyltransferases (HMTs) constitute a large family that includes histone lysine methyltransferases (HKMTs) and histone/protein arginine methyltransferases (PRMTs). There is increasing evidence showing a correlation between HKMTs and cancer pathogenesis. Here, we present an overview of representative HKMTs, including their biological and biochemical properties as well as the profiles of small molecule inhibitors for a comprehensive understanding of HKMTs in drug discovery. Keywords: histone methyltransferases; adenosylmethionine; small molecule inhibitors; anti-cancer drugs; epigenetic modification; drug discovery Acta Pharmacologica Sinica (2016) 37: 1273–1280; doi: 10.1038/aps.2016.64; published online 11 Jul 2016 Introduction vivo, suggesting the possibility of using HKMTs as targets for Post-translational epigenetic modifications of histones are cancer therapy. Here, we discuss individual histone lysine controlled by histone-modifying enzymes, including histone methylation with the currently available small molecule inhib- methyltransferases (HMTs), histone demethylases, histone itors and their applications in cancer treatment. acetyltransferases, histone deacetylases (HDACs), ubiquitin ligases, and other specific kinases phosphorylating serine HKMTs-catalyzed methylation residues on histones.