An Atlas of Chromatin Accessibility in the Adult Human Brain
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Analysis of the Calcineurin Pathway Identifies Members of the EGR Gene Family, Specifically EGR3, As Potential Susceptibility Candidates in Schizophrenia
Genetic analysis of the calcineurin pathway identifies members of the EGR gene family, specifically EGR3, as potential susceptibility candidates in schizophrenia Kazuo Yamada*, David J. Gerber†, Yoshimi Iwayama*, Tetsuo Ohnishi*, Hisako Ohba*, Tomoko Toyota*, Jun Aruga‡, Yoshio Minabe*§, Susumu Tonegawa†¶, and Takeo Yoshikawa*ʈ** Laboratories for *Molecular Psychiatry and ‡Comparative Neural Development, RIKEN Brain Science Institute, Saitama 351-0198, Japan; †Howard Hughes Medical Institute and RIKEN–MIT Neuroscience Research Center, The Picower Institute for Learning and Memory, Massachusetts Institute of Technology, Cambridge, MA 02139; §Department of Psychiatry and Neurology, Kanazawa University School of Medicine, Ishikawa 920-8641, Japan; and ʈCore Research for Evolutional Science and Technology, Japan Science and Technology Agency, Saitama 332-0012, Japan Contributed by Susumu Tonegawa, December 7, 2006 (sent for review September 22, 2006) The calcineurin cascade is central to neuronal signal transduction, cineurin is particularly enriched in the CNS, where it plays a critical and genes in this network are intriguing candidate schizophrenia role in the regulation of a diverse array of neuronal functions (5, 6). susceptibility genes. To replicate and extend our previously re- Interestingly, calcineurin is positioned downstream of dopaminer- ported association between the PPP3CC gene, encoding the cal- gic signaling (7) and is involved in NMDA receptor-mediated cineurin catalytic ␥-subunit, and schizophrenia, we examined 84 synaptic plasticity (8) and could therefore provide an important SNPs from 14 calcineurin-related candidate genes for genetic as- functional link between these two neurotransmitter systems. To sociation by using 124 Japanese schizophrenic pedigrees. Four of further explore the involvement of calcineurin dysfunction in these genes (PPP3CC, EGR2, EGR3, and EGR4) showed nominally schizophrenia, we have tested for genetic association of a subset of significant association with schizophrenia. -

Genazzani-2003-Calcineurin And
Edinburgh Research Explorer Calcineurin controls the expression of numerous genes in cerebellar granule cells Citation for published version: Kramer, D, Fresu, L, Ashby, DS, Freeman, TC & Genazzani, AA 2003, 'Calcineurin controls the expression of numerous genes in cerebellar granule cells', Molecular and Cellular Neuroscience, vol. 23, no. 2, pp. 325- 30. Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: Molecular and Cellular Neuroscience Publisher Rights Statement: © 2003 Elsevier Science (USA) General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 28. Sep. 2021 Molecular and Cellular Neuroscience 23 (2003) 325–330 www.elsevier.com/locate/ymcne Calcineurin controls the expression of numerous genes in cerebellar granule cells Dana Kramer,a Luigia Fresu,b Dominique S. Ashby,a Tom C. Freeman,c and Armando A. Genazzania,* a Department of Pharmacology, Tennis Court Road, Cambridge, CB2 1PD, UK b Department of Pharmacology, Gynaecology and Obstetrics, University of Sassari, Italy c Microarray Group, MRC HGMP-RC, Wellcome Trust Genome Campus, Hinxton, CB10 1SB, UK Received 31 October 2002; revised 7 January 2003; accepted 20 January 2003 Abstract The Ca2ϩ/calmodulin-dependent phosphatase calcineurin plays a crucial role in gene expression in different cell types such as T-lymphocytes, cardiac myocytes, and smooth muscle cells. -

Viewed Under 23 (B) Or 203 (C) fi M M Male Cko Mice, and Largely Unaffected Magni Cation; Scale Bars, 500 M (B) and 50 M (C)
BRIEF COMMUNICATION www.jasn.org Renal Fanconi Syndrome and Hypophosphatemic Rickets in the Absence of Xenotropic and Polytropic Retroviral Receptor in the Nephron Camille Ansermet,* Matthias B. Moor,* Gabriel Centeno,* Muriel Auberson,* † † ‡ Dorothy Zhang Hu, Roland Baron, Svetlana Nikolaeva,* Barbara Haenzi,* | Natalya Katanaeva,* Ivan Gautschi,* Vladimir Katanaev,*§ Samuel Rotman, Robert Koesters,¶ †† Laurent Schild,* Sylvain Pradervand,** Olivier Bonny,* and Dmitri Firsov* BRIEF COMMUNICATION *Department of Pharmacology and Toxicology and **Genomic Technologies Facility, University of Lausanne, Lausanne, Switzerland; †Department of Oral Medicine, Infection, and Immunity, Harvard School of Dental Medicine, Boston, Massachusetts; ‡Institute of Evolutionary Physiology and Biochemistry, St. Petersburg, Russia; §School of Biomedicine, Far Eastern Federal University, Vladivostok, Russia; |Services of Pathology and ††Nephrology, Department of Medicine, University Hospital of Lausanne, Lausanne, Switzerland; and ¶Université Pierre et Marie Curie, Paris, France ABSTRACT Tight control of extracellular and intracellular inorganic phosphate (Pi) levels is crit- leaves.4 Most recently, Legati et al. have ical to most biochemical and physiologic processes. Urinary Pi is freely filtered at the shown an association between genetic kidney glomerulus and is reabsorbed in the renal tubule by the action of the apical polymorphisms in Xpr1 and primary fa- sodium-dependent phosphate transporters, NaPi-IIa/NaPi-IIc/Pit2. However, the milial brain calcification disorder.5 How- molecular identity of the protein(s) participating in the basolateral Pi efflux remains ever, the role of XPR1 in the maintenance unknown. Evidence has suggested that xenotropic and polytropic retroviral recep- of Pi homeostasis remains unknown. Here, tor 1 (XPR1) might be involved in this process. Here, we show that conditional in- we addressed this issue in mice deficient for activation of Xpr1 in the renal tubule in mice resulted in impaired renal Pi Xpr1 in the nephron. -

Egr-1 Induces DARPP-32 Expression in Striatal Medium Spiny Neurons Via a Conserved Intragenic Element
6808 • The Journal of Neuroscience, May 16, 2012 • 32(20):6808–6818 Cellular/Molecular Egr-1 Induces DARPP-32 Expression in Striatal Medium Spiny Neurons via a Conserved Intragenic Element Serene Keilani,1 Samira Chandwani,1 Georgia Dolios,2 Alexey Bogush,3 Heike Beck,4 Antonis K. Hatzopoulos,5 Gadiparthi N. Rao,6 Elizabeth A. Thomas,7 Rong Wang,2 and Michelle E. Ehrlich1,2 Departments of 1Neurology and Pediatrics and 2Genetics and Genomic Sciences, Mount Sinai School of Medicine, New York, New York 10029, 3Weinberg Unit for ALS Research, Department of Neuroscience, Farber Institute for Neuroscience, Thomas Jefferson University, Philadelphia, Pennsylvania 19107, 4Walter Brendel Center of Experimental Medicine, Ludwig Maximilians University, D-81377 Munich, Germany, 5Division of Cardiovascular Medicine, Department of Medicine, Vanderbilt University, Nashville, Tennessee 37240, 6Department of Physiology, University of Tennessee Health Science Center, Memphis, Tennessee 38163, and 7Department of Molecular Biology, The Scripps Research Institute, La Jolla, California 92037 DARPP-32 (dopamine and adenosine 3Ј,5Ј-cyclic monophosphate cAMP-regulated phosphoprotein, 32 kDa) is a striatal-enriched protein that mediates signaling by dopamine and other first messengers in the medium spiny neurons. The transcriptional mechanisms that regulate striatal DARPP-32 expression remain enigmatic and are a subject of much interest in the efforts to induce a striatal phenotype in stem cells. We report the identification and characterization of a conserved region, also known as H10, in intron IV of the gene that codes for DARPP-32 (Ppp1r1b). This DNA sequence forms multiunit complexes with nuclear proteins from adult and embry- onic striata of mice and rats. -

Psychostimulant-Regulated Plasticity in Interneurons of the Nucleus Accumbens
Psychostimulant-Regulated Plasticity in Interneurons of the Nucleus Accumbens by David A. Gallegos Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Anne E. West, Supervisor ___________________________ Jorg Grandl ___________________________ Debra Silver ___________________________ Gregory Crawford ___________________________ Hiro Matsunami Psychostimulant-Regulated Epigenetic Plasticity in Interneurons of the Nucleus Accumbens submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2019 ABSTRACT Psychostimulant-Regulated Epigenetic Plasticity in Interneurons of the Nucleus Accumbens by David A. Gallegos Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Anne E. West, Supervisor ___________________________ Jorg Grandl ___________________________ Debra Silver ___________________________ Gregory Crawford ___________________________ Hiro Matsunami An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2019 Copyright by David Andres Gallegos 2019 Abstract Exposure to psychostimulant drugs of abuse exerts lasting influences on brain function via the regulation of immediate and persistent gene transcription. These changes in gene transcription drive the development of addictive-like -

DRD2 and PPP1R1B (DARPP-32)
Hettinger et al. Behavioral and Brain Functions 2012, 8:19 http://www.behavioralandbrainfunctions.com/8/1/19 RESEARCH Open Access DRD2 and PPP1R1B (DARPP-32) polymorphisms independently confer increased risk for autism spectrum disorders and additively predict affected status in male-only affected sib-pair families Joe A Hettinger1, Xudong Liu2,3, Melissa L Hudson2,3,4, Alana Lee2,3,4, Ira L Cohen4,5, Ron C Michaelis6, Charles E Schwartz7, Suzanne ME Lewis4,8,9 and Jeanette JA Holden1,2,3,4,10,11* Abstract Background: The neurotransmitter dopamine (DA) modulates executive functions, learning, and emotional processing, all of which are impaired in individuals with autism spectrum disorders (ASDs). Our previous findings suggest a role for dopamine-related genes in families with only affected males. Methods: We examined two additional genes which affect DA function, the DRD2 and PPP1R1B (DARPP-32) genes, in a cohort of 112 male-only affected sib-pair families. Selected polymorphisms spanning these genes were genotyped and both family-based and population-based tests were carried out for association analysis. General discriminant analysis was used to examine the gene-gene interactions in predicting autism susceptibility. Results: There was a significantly increased frequency of the DRD2 rs1800498TT genotype (P = 0.007) in affected males compared to the comparison group, apparently due to over-transmission of the T allele (P = 0.0003). The frequency of the PPP1R1B rs1495099CC genotype in affected males was also higher than that in the comparison group (P = 0.002) due to preferential transmission of the C allele from parents to affected children (P = 0.0009). -

Effects of PPP1R1B (DARPP-32) Polymorphism on Feedback-Related Brain Potentials Across the Life Span
ORIGINAL RESEARCH ARTICLE published: 04 March 2013 doi: 10.3389/fpsyg.2013.00089 Effects of PPP1R1B (DARPP-32) polymorphism on feedback-related brain potentials across the life span Dorothea Hämmerer 1,2*†, Gudio Biele 3,4,Viktor Müller 1, HolgerThiele 5, Peter Nürnberg 5,6,7, Hauke R. Heekeren1,3 and Shu-Chen Li 1,2*† 1 Center for Lifespan Psychology, Max Planck Institute for Human Development, Berlin, Germany 2 Department of Psychology, Technische Universität Dresden, Dresden, Germany 3 Department of Education and Psychology, Freie Universität Berlin, Berlin, Germany 4 Institute of Psychology, University of Oslo, Oslo, Norway 5 Cologne Center for Genomics, University of Cologne, Cologne, Germany 6 Center for Molecular Medicine Cologne, University of Cologne, Cologne, Germany 7 Cologne Excellence Cluster on Cellular Stress Responses in Aging-Associated Diseases, University of Cologne, Cologne, Germany Edited by: Maximizing gains during probabilistic reinforcement learning requires the updating of Alain Dagher, Montreal Neurological choice – outcome expectations at the time when the feedback about a specific choice Institute and Hospital, Canada or action is given. Extant theories and evidence suggest that dopaminergic modulation Reviewed by: Meghana Bhatt, City of Hope, USA plays a crucial role in reinforcement learning and the updating of choice – outcome expec- James F.Cavanagh, Brown University, tations. Furthermore, recently a positive component of the event-related potential about USA 200 ms (P2) after feedback has been suggested to reflect such updating. The efficacy of *Correspondence: dopaminergic modulation changes across the life span. However, to date investigations Dorothea Hämmerer and Shu-Chen of age-related differences in feedback-related P2 during reinforcement learning are still Li, Department of Psychology, Technische Universität Dresden, scarce. -
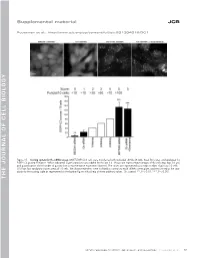
Thejournalofcellb Io Logy
Supplemental material JCB Puustinen et al., http://www.jcb.org/cgi/content/full/jcb.201304012/DC1 Figure S1. Scoring system for the siRNA screen. MCF7-EGFP-LC3 cells were transfected with indicated siRNAs (8 nM), fixed 56 h later, and analyzed for EGFP-LC3 puncta formation. When indicated, 5 µM siramesine was added for the last 3 h. Shown are representative images of the cells (top; bar, 10 µm) and quantification of the number of puncta from a representative experiment (bottom). The values are represented as a mean number of puncta/10 cells ± SDs from four randomly chosen areas of 10 cells. The shown treatments were included as controls to each siRNA screen plate, and they served as the stan- dards for the scoring scale as represented on the bottom figure with a help of three arbitrary values. Ctr, control. **, P < 0.01; ***, P < 0.001. THE JOURNAL OF CELL BIOLOGY CIP2A regulates mTORC1, cell growth, and autophagy • Puustinen et al. S1 Figure S2. A cartoon highlighting the positions of the candidate autophagy-regulating genes in the insulin receptor signaling network. The cartoon was prepared by Ingenuity Pathway Analysis. software. CP, carbohydrate phosphatase; ERK, extracellular signal–regulated kinase; FSH, follicle-stimulating hor- mone; EG, epidermal growth factor. S2 JCB Figure S3. The effect of siRNAs targeting the autophagy-regulating PP2 subunits on target gene expression, cell density, and mTORC1 pathway. (A) Total RNA was isolated from MCF7 cells treated with indicated siRNAs (20 nM) for 54 h and analyzed for target gene expression by qPCR using primers de- signed to specifically amplify the indicated PP2A-related mRNAs andACTB mRNA (internal control). -

Dopamine Response Gene Pathways in Dorsal Striatum Msns from a Gene
bioRxiv preprint doi: https://doi.org/10.1101/757500; this version posted September 5, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Dopamine response gene pathways in dorsal striatum MSNs from a gene 2 expression viewpoint: cAMP-mediated gene networks 3 Vladimir Babenko, Institute of Cytology and Genetics SB RAS, Novosibirsk, Russia 630090, Novosibirsk 4 State University, Novosibirsk, Russia 630090; Anna Galyamina, Institute of Cytology and Genetics SB RAS, 5 Novosibirsk, Russia 630090; Igor Rogozin, National Institutes of Health, Rockville Pike, Bethesda, Maryland, 6 USA 20892; Dmitry Smagin, Institute of Cytology and Genetics SB RAS, Novosibirsk, Russia 630090; 7 Natalia Kudryavtseva, Institute of Cytology and Genetics SB RAS, Novosibirsk, Russia 630090 8 9 *Corresponding author e-mail address: [email protected] (Vladimir Babenko) 10 Abstract 11 A mouse model of chronic social conflicts was used to analyze dorsal striatum neurons implicated in 12 cAMP-mediated phosphorylation activation pathways specific for Medium Spiny Neurons (MSNs). Based on 13 expression correlation analysis, we succeeded in dissecting Drd1- and Drd2-dopaminoceptive neurons (D1 14 and D2, correspondingly) gene pathways. We also found that D1 neurons feature previously reported two 15 states, passive and active ones, represented in our analysis by distinct, negatively correlated gene clusters. 16 The correlation based gene pathways strongly corroborate the phosphorylation cascades highlighted 17 in the previous studies, implying that the expression-based viewpoint corresponds to phosphorylation/ 18 dephosphorylation interplay in each type of neurons. Notably, D2 neurons showed the largest Ppp1r1b 19 (encoding DARPP-32) expression modulation impact, implying that Ppp1r1b expression dynamics is mostly 20 associated with neuroendocrine response mediated by Penk/Pdyn genes expression in D2 neurons. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Genome-Wide Identification of Brain Mirnas in Response to High
Zhao et al. BMC Molecular Biol (2019) 20:3 https://doi.org/10.1186/s12867-019-0120-4 BMC Molecular Biology RESEARCH ARTICLE Open Access Genome‑wide identifcation of brain miRNAs in response to high‑intensity intermittent swimming training in Rattus norvegicus by deep sequencing Yanhong Zhao1*†, Anmin Zhang2,3*†, Yanfang Wang4, Shuping Hu3, Ruiping Zhang2 and Shuaiwei Qian2 Abstract Background: Physical exercise can improve brain function by altering brain gene expression. The expression mecha- nisms underlying the brain’s response to exercise still remain unknown. miRNAs as vital regulators of gene expres- sion may be involved in regulation of brain genes in response to exercise. However, as yet, very little is known about exercise-responsive miRNAs in brain. Results: We constructed two comparative small RNA libraries of rat brain from a high-intensity intermittent swim- ming training (HIST) group and a normal control (NC) group. Using deep sequencing and bioinformatics analysis, we identifed 2109 (1700 from HIST, 1691 from NC) known and 55 (50 from HIST, 28 from NC) novel candidate miRNAs. Among them, 34 miRNAs were identifed as signifcantly diferentially expressed in response to HIST, 16 were up- regulated and 18 were down-regulated. The results showed that all members of mir-200 family were strongly up- regulated, implying mir-200 family may play very important roles in HIST response mechanisms of rat brain. A total of 955 potential target genes of these 34 exercise-responsive miRNAs were identifed from rat genes. Most of them are directly involved in the development and regulatory function of brain or nerve. -

Comparison of Gene Expression Profiles Between Dental Pulp and Periodontal Ligament Tissues in Humans
INTERNATIONAL JOURNAL OF MOLECULAR MEDICINE 40: 647-660, 2017 Comparison of gene expression profiles between dental pulp and periodontal ligament tissues in humans AI-XIU GONG1*, JING-HAN ZHANG2*, JING LI1, JUN WU3, LIN WANG4 and DENG-SHUN MIAO3 Departments of 1Stomatology, and 2Neonatology, Children's Hospital of Nanjing Medical University, Nanjing, Jiangsu 210008; 3State Key Laboratory of Reproductive Medicine, The Research Center for Bone and Stem Cells, Department of Anatomy, Histology and Embryology, Nanjing Medical University; 4Jiangsu Key Laboratory of Oral Diseases, Department of Orthodontics, School of Stomatology, Nanjing Medical University, Nanjing, Jiangsu 210029, P.R. China Received March 16, 2016; Accepted June 16, 2017 DOI: 10.3892/ijmm.2017.3065 Abstract. There are anatomical and functional differ- DP samples. The gene expression profiles provide novel insight ences between human dental pulp (DP) and periodontal into the characterization of DP and PDL tissues, and contribute ligament (PDL). However, the molecular biological differ- to our understanding of the potential molecular mechanisms of ences and function of these tissues are poorly understood. dental tissue mineralization and regeneration. In the present study, we employed a cDNA microarray array to screen for differentially expressed genes (DEGs) between Introduction human DP and PDL tissues, and used the online software WebGestalt to perform the functional analysis of the DEGs. Dental pulp (DP) tissue, termed as the ‘ectomesenchyme’, is In addition, the STRING database and KEGG pathway derived from ectodermal cells that grow on the periphery of analysis were applied for interaction network and pathway the neural tube, migrate to the oral position and then differen- analysis of the DEGs.