UNIVERSITY of CALIFORNIA, IRVINE Epigenetic Control of Medial Habenula Function in Cocaine-Associated Behaviors DISSERTATION
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Analysis of the Calcineurin Pathway Identifies Members of the EGR Gene Family, Specifically EGR3, As Potential Susceptibility Candidates in Schizophrenia
Genetic analysis of the calcineurin pathway identifies members of the EGR gene family, specifically EGR3, as potential susceptibility candidates in schizophrenia Kazuo Yamada*, David J. Gerber†, Yoshimi Iwayama*, Tetsuo Ohnishi*, Hisako Ohba*, Tomoko Toyota*, Jun Aruga‡, Yoshio Minabe*§, Susumu Tonegawa†¶, and Takeo Yoshikawa*ʈ** Laboratories for *Molecular Psychiatry and ‡Comparative Neural Development, RIKEN Brain Science Institute, Saitama 351-0198, Japan; †Howard Hughes Medical Institute and RIKEN–MIT Neuroscience Research Center, The Picower Institute for Learning and Memory, Massachusetts Institute of Technology, Cambridge, MA 02139; §Department of Psychiatry and Neurology, Kanazawa University School of Medicine, Ishikawa 920-8641, Japan; and ʈCore Research for Evolutional Science and Technology, Japan Science and Technology Agency, Saitama 332-0012, Japan Contributed by Susumu Tonegawa, December 7, 2006 (sent for review September 22, 2006) The calcineurin cascade is central to neuronal signal transduction, cineurin is particularly enriched in the CNS, where it plays a critical and genes in this network are intriguing candidate schizophrenia role in the regulation of a diverse array of neuronal functions (5, 6). susceptibility genes. To replicate and extend our previously re- Interestingly, calcineurin is positioned downstream of dopaminer- ported association between the PPP3CC gene, encoding the cal- gic signaling (7) and is involved in NMDA receptor-mediated cineurin catalytic ␥-subunit, and schizophrenia, we examined 84 synaptic plasticity (8) and could therefore provide an important SNPs from 14 calcineurin-related candidate genes for genetic as- functional link between these two neurotransmitter systems. To sociation by using 124 Japanese schizophrenic pedigrees. Four of further explore the involvement of calcineurin dysfunction in these genes (PPP3CC, EGR2, EGR3, and EGR4) showed nominally schizophrenia, we have tested for genetic association of a subset of significant association with schizophrenia. -

Egr-1 Induces DARPP-32 Expression in Striatal Medium Spiny Neurons Via a Conserved Intragenic Element
6808 • The Journal of Neuroscience, May 16, 2012 • 32(20):6808–6818 Cellular/Molecular Egr-1 Induces DARPP-32 Expression in Striatal Medium Spiny Neurons via a Conserved Intragenic Element Serene Keilani,1 Samira Chandwani,1 Georgia Dolios,2 Alexey Bogush,3 Heike Beck,4 Antonis K. Hatzopoulos,5 Gadiparthi N. Rao,6 Elizabeth A. Thomas,7 Rong Wang,2 and Michelle E. Ehrlich1,2 Departments of 1Neurology and Pediatrics and 2Genetics and Genomic Sciences, Mount Sinai School of Medicine, New York, New York 10029, 3Weinberg Unit for ALS Research, Department of Neuroscience, Farber Institute for Neuroscience, Thomas Jefferson University, Philadelphia, Pennsylvania 19107, 4Walter Brendel Center of Experimental Medicine, Ludwig Maximilians University, D-81377 Munich, Germany, 5Division of Cardiovascular Medicine, Department of Medicine, Vanderbilt University, Nashville, Tennessee 37240, 6Department of Physiology, University of Tennessee Health Science Center, Memphis, Tennessee 38163, and 7Department of Molecular Biology, The Scripps Research Institute, La Jolla, California 92037 DARPP-32 (dopamine and adenosine 3Ј,5Ј-cyclic monophosphate cAMP-regulated phosphoprotein, 32 kDa) is a striatal-enriched protein that mediates signaling by dopamine and other first messengers in the medium spiny neurons. The transcriptional mechanisms that regulate striatal DARPP-32 expression remain enigmatic and are a subject of much interest in the efforts to induce a striatal phenotype in stem cells. We report the identification and characterization of a conserved region, also known as H10, in intron IV of the gene that codes for DARPP-32 (Ppp1r1b). This DNA sequence forms multiunit complexes with nuclear proteins from adult and embry- onic striata of mice and rats. -

Psychostimulant-Regulated Plasticity in Interneurons of the Nucleus Accumbens
Psychostimulant-Regulated Plasticity in Interneurons of the Nucleus Accumbens by David A. Gallegos Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Anne E. West, Supervisor ___________________________ Jorg Grandl ___________________________ Debra Silver ___________________________ Gregory Crawford ___________________________ Hiro Matsunami Psychostimulant-Regulated Epigenetic Plasticity in Interneurons of the Nucleus Accumbens submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2019 ABSTRACT Psychostimulant-Regulated Epigenetic Plasticity in Interneurons of the Nucleus Accumbens by David A. Gallegos Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Anne E. West, Supervisor ___________________________ Jorg Grandl ___________________________ Debra Silver ___________________________ Gregory Crawford ___________________________ Hiro Matsunami An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2019 Copyright by David Andres Gallegos 2019 Abstract Exposure to psychostimulant drugs of abuse exerts lasting influences on brain function via the regulation of immediate and persistent gene transcription. These changes in gene transcription drive the development of addictive-like -

DRD2 and PPP1R1B (DARPP-32)
Hettinger et al. Behavioral and Brain Functions 2012, 8:19 http://www.behavioralandbrainfunctions.com/8/1/19 RESEARCH Open Access DRD2 and PPP1R1B (DARPP-32) polymorphisms independently confer increased risk for autism spectrum disorders and additively predict affected status in male-only affected sib-pair families Joe A Hettinger1, Xudong Liu2,3, Melissa L Hudson2,3,4, Alana Lee2,3,4, Ira L Cohen4,5, Ron C Michaelis6, Charles E Schwartz7, Suzanne ME Lewis4,8,9 and Jeanette JA Holden1,2,3,4,10,11* Abstract Background: The neurotransmitter dopamine (DA) modulates executive functions, learning, and emotional processing, all of which are impaired in individuals with autism spectrum disorders (ASDs). Our previous findings suggest a role for dopamine-related genes in families with only affected males. Methods: We examined two additional genes which affect DA function, the DRD2 and PPP1R1B (DARPP-32) genes, in a cohort of 112 male-only affected sib-pair families. Selected polymorphisms spanning these genes were genotyped and both family-based and population-based tests were carried out for association analysis. General discriminant analysis was used to examine the gene-gene interactions in predicting autism susceptibility. Results: There was a significantly increased frequency of the DRD2 rs1800498TT genotype (P = 0.007) in affected males compared to the comparison group, apparently due to over-transmission of the T allele (P = 0.0003). The frequency of the PPP1R1B rs1495099CC genotype in affected males was also higher than that in the comparison group (P = 0.002) due to preferential transmission of the C allele from parents to affected children (P = 0.0009). -

Effects of PPP1R1B (DARPP-32) Polymorphism on Feedback-Related Brain Potentials Across the Life Span
ORIGINAL RESEARCH ARTICLE published: 04 March 2013 doi: 10.3389/fpsyg.2013.00089 Effects of PPP1R1B (DARPP-32) polymorphism on feedback-related brain potentials across the life span Dorothea Hämmerer 1,2*†, Gudio Biele 3,4,Viktor Müller 1, HolgerThiele 5, Peter Nürnberg 5,6,7, Hauke R. Heekeren1,3 and Shu-Chen Li 1,2*† 1 Center for Lifespan Psychology, Max Planck Institute for Human Development, Berlin, Germany 2 Department of Psychology, Technische Universität Dresden, Dresden, Germany 3 Department of Education and Psychology, Freie Universität Berlin, Berlin, Germany 4 Institute of Psychology, University of Oslo, Oslo, Norway 5 Cologne Center for Genomics, University of Cologne, Cologne, Germany 6 Center for Molecular Medicine Cologne, University of Cologne, Cologne, Germany 7 Cologne Excellence Cluster on Cellular Stress Responses in Aging-Associated Diseases, University of Cologne, Cologne, Germany Edited by: Maximizing gains during probabilistic reinforcement learning requires the updating of Alain Dagher, Montreal Neurological choice – outcome expectations at the time when the feedback about a specific choice Institute and Hospital, Canada or action is given. Extant theories and evidence suggest that dopaminergic modulation Reviewed by: Meghana Bhatt, City of Hope, USA plays a crucial role in reinforcement learning and the updating of choice – outcome expec- James F.Cavanagh, Brown University, tations. Furthermore, recently a positive component of the event-related potential about USA 200 ms (P2) after feedback has been suggested to reflect such updating. The efficacy of *Correspondence: dopaminergic modulation changes across the life span. However, to date investigations Dorothea Hämmerer and Shu-Chen of age-related differences in feedback-related P2 during reinforcement learning are still Li, Department of Psychology, Technische Universität Dresden, scarce. -
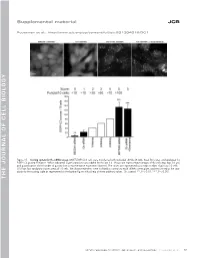
Thejournalofcellb Io Logy
Supplemental material JCB Puustinen et al., http://www.jcb.org/cgi/content/full/jcb.201304012/DC1 Figure S1. Scoring system for the siRNA screen. MCF7-EGFP-LC3 cells were transfected with indicated siRNAs (8 nM), fixed 56 h later, and analyzed for EGFP-LC3 puncta formation. When indicated, 5 µM siramesine was added for the last 3 h. Shown are representative images of the cells (top; bar, 10 µm) and quantification of the number of puncta from a representative experiment (bottom). The values are represented as a mean number of puncta/10 cells ± SDs from four randomly chosen areas of 10 cells. The shown treatments were included as controls to each siRNA screen plate, and they served as the stan- dards for the scoring scale as represented on the bottom figure with a help of three arbitrary values. Ctr, control. **, P < 0.01; ***, P < 0.001. THE JOURNAL OF CELL BIOLOGY CIP2A regulates mTORC1, cell growth, and autophagy • Puustinen et al. S1 Figure S2. A cartoon highlighting the positions of the candidate autophagy-regulating genes in the insulin receptor signaling network. The cartoon was prepared by Ingenuity Pathway Analysis. software. CP, carbohydrate phosphatase; ERK, extracellular signal–regulated kinase; FSH, follicle-stimulating hor- mone; EG, epidermal growth factor. S2 JCB Figure S3. The effect of siRNAs targeting the autophagy-regulating PP2 subunits on target gene expression, cell density, and mTORC1 pathway. (A) Total RNA was isolated from MCF7 cells treated with indicated siRNAs (20 nM) for 54 h and analyzed for target gene expression by qPCR using primers de- signed to specifically amplify the indicated PP2A-related mRNAs andACTB mRNA (internal control). -

Dopamine Response Gene Pathways in Dorsal Striatum Msns from a Gene
bioRxiv preprint doi: https://doi.org/10.1101/757500; this version posted September 5, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Dopamine response gene pathways in dorsal striatum MSNs from a gene 2 expression viewpoint: cAMP-mediated gene networks 3 Vladimir Babenko, Institute of Cytology and Genetics SB RAS, Novosibirsk, Russia 630090, Novosibirsk 4 State University, Novosibirsk, Russia 630090; Anna Galyamina, Institute of Cytology and Genetics SB RAS, 5 Novosibirsk, Russia 630090; Igor Rogozin, National Institutes of Health, Rockville Pike, Bethesda, Maryland, 6 USA 20892; Dmitry Smagin, Institute of Cytology and Genetics SB RAS, Novosibirsk, Russia 630090; 7 Natalia Kudryavtseva, Institute of Cytology and Genetics SB RAS, Novosibirsk, Russia 630090 8 9 *Corresponding author e-mail address: [email protected] (Vladimir Babenko) 10 Abstract 11 A mouse model of chronic social conflicts was used to analyze dorsal striatum neurons implicated in 12 cAMP-mediated phosphorylation activation pathways specific for Medium Spiny Neurons (MSNs). Based on 13 expression correlation analysis, we succeeded in dissecting Drd1- and Drd2-dopaminoceptive neurons (D1 14 and D2, correspondingly) gene pathways. We also found that D1 neurons feature previously reported two 15 states, passive and active ones, represented in our analysis by distinct, negatively correlated gene clusters. 16 The correlation based gene pathways strongly corroborate the phosphorylation cascades highlighted 17 in the previous studies, implying that the expression-based viewpoint corresponds to phosphorylation/ 18 dephosphorylation interplay in each type of neurons. Notably, D2 neurons showed the largest Ppp1r1b 19 (encoding DARPP-32) expression modulation impact, implying that Ppp1r1b expression dynamics is mostly 20 associated with neuroendocrine response mediated by Penk/Pdyn genes expression in D2 neurons. -

Genome-Wide Identification of Brain Mirnas in Response to High
Zhao et al. BMC Molecular Biol (2019) 20:3 https://doi.org/10.1186/s12867-019-0120-4 BMC Molecular Biology RESEARCH ARTICLE Open Access Genome‑wide identifcation of brain miRNAs in response to high‑intensity intermittent swimming training in Rattus norvegicus by deep sequencing Yanhong Zhao1*†, Anmin Zhang2,3*†, Yanfang Wang4, Shuping Hu3, Ruiping Zhang2 and Shuaiwei Qian2 Abstract Background: Physical exercise can improve brain function by altering brain gene expression. The expression mecha- nisms underlying the brain’s response to exercise still remain unknown. miRNAs as vital regulators of gene expres- sion may be involved in regulation of brain genes in response to exercise. However, as yet, very little is known about exercise-responsive miRNAs in brain. Results: We constructed two comparative small RNA libraries of rat brain from a high-intensity intermittent swim- ming training (HIST) group and a normal control (NC) group. Using deep sequencing and bioinformatics analysis, we identifed 2109 (1700 from HIST, 1691 from NC) known and 55 (50 from HIST, 28 from NC) novel candidate miRNAs. Among them, 34 miRNAs were identifed as signifcantly diferentially expressed in response to HIST, 16 were up- regulated and 18 were down-regulated. The results showed that all members of mir-200 family were strongly up- regulated, implying mir-200 family may play very important roles in HIST response mechanisms of rat brain. A total of 955 potential target genes of these 34 exercise-responsive miRNAs were identifed from rat genes. Most of them are directly involved in the development and regulatory function of brain or nerve. -

Dopamine Response Gene Pathways in Dorsal Striatum Msns from a Gene Expression Viewpoint: Camp‑Mediated Gene Networks Vladimir N
Babenko et al. BMC Neurosci (2020) 21:12 https://doi.org/10.1186/s12868-020-00560-w BMC Neuroscience RESEARCH ARTICLE Open Access Dopamine response gene pathways in dorsal striatum MSNs from a gene expression viewpoint: cAMP-mediated gene networks Vladimir N. Babenko1,2*, Anna G. Galyamina1, Igor B. Rogozin3, Dmitry A. Smagin1 and Natalia N. Kudryavtseva1 Abstract Background: Medium spiny neurons (MSNs) comprise the main body (95% in mouse) of the dorsal striatum neu- rons and represent dopaminoceptive GABAergic neurons. The cAMP (cyclic Adenosine MonoPhosphate)—mediated cascade of excitation and inhibition responses observed in MSN intracellular signal transduction is crucial for neuro- science research due to its involvement in the motor and behavioral functions. In particular, all types of addictions are related to MSNs. Shedding the light on the mechanics of the above-mentioned cascade is of primary importance for this research domain. Results: A mouse model of chronic social conficts in daily agonistic interactions was used to analyze dorsal stria- tum neurons genes implicated in cAMP-mediated phosphorylation activation pathways specifc for MSNs. Based on expression correlation analysis, we succeeded in dissecting Drd1- and Drd2-dopaminoceptive neurons (D1 and D2, correspondingly) gene pathways. We also found that D1 neurons genes clustering are split into two oppositely cor- related states, passive and active ones, the latter apparently corresponding to D1 fring stage upon protein kinase A (PKA) activation. We observed that under defeat stress in chronic social conficts the loser mice manifest overall depression of dopa- mine-mediated MSNs activity resulting in previously reported reduced motor activity, while the aggressive mice with positive fghting experience (aggressive mice) feature an increase in both D1-active phase and D2 MSNs genes expression leading to hyperactive behavior pattern corresponded by us before. -

Research Paper: Genetic Variations of DAOA (Rs947267 and Rs3918342) and COMT Genes (Rs165599 and Rs4680) in Schizophrenia and Bipolar I Disorder
Basic and Clinical November, December 2018, Volume 9, Number 6 Research Paper: Genetic Variations of DAOA (rs947267 and rs3918342) and COMT Genes (rs165599 and rs4680) in Schizophrenia and Bipolar I Disorder Leila Ahmadi1 , Seyed Reza Kazemi Nezhad1* , Parisima Behbahani1 , Nilofar Khajeddin2 , Mehdi Pourmehdi-Boroujeni3 1. Department of Genetics, Faculty of Sciences, Shahid Chamran University of Ahvaz, Ahvaz, Iran. 2. Department of Psychiatry, School of Medicine, Ahvaz Jundishapur University of Medical Sciences, Ahvaz, Iran. 3. Department of Food Hygiene, Faculty of Veterinary Medicine, Shahid Chamran University of Ahvaz, Ahvaz, Iran. Use your device to scan and read the article online Citation: Ahmadi, L., Kazemi Nezhad, S. R., Behbahani, P., Khajeddin, N., & Pourmehdi-Boroujeni. M. (2018). Genetic Variations of DAOA (rs947267 and rs3918342) and COMT Genes (rs165599 and rs4680) in Schizophrenia and Bipolar I Disorder. Basic and Clinical Neuroscience, 9(6), 429-438. http://dx.doi.org/10.32598/bcn.9.6.429 : http://dx.doi.org/10.32598/bcn.9.6.429 A B S T R A C T Funding: See Page 436 Introduction: Genetic and environmental factors are involved in the incidence of schizophrenia and bipolar disorder. Many reports confirm that several common genes are connected with Article info: these two psychotic disorders. Several neurotransmitters may be involved in the molecular Received: 01 February 2017 mechanisms of schizophrenia and bipolar disorder. We aimed to estimate the role of two First Revision: 10 March 2017 talent genes: DAOA in neurotransmission of glutamate and COMT in neurotransmission of Accepted: 26 May 2018 dopamine to guide the treatment of schizophrenia and bipolar disorder. -

An Atlas of Chromatin Accessibility in the Adult Human Brain
Downloaded from genome.cshlp.org on October 4, 2021 - Published by Cold Spring Harbor Laboratory Press Resource An atlas of chromatin accessibility in the adult human brain John F. Fullard,1,2,3,12 Mads E. Hauberg,1,2,4,5,6,12 Jaroslav Bendl,1,2,3 Gabor Egervari,1,2,7 Maria-Daniela Cirnaru,8 Sarah M. Reach,3 Jan Motl,9 Michelle E. Ehrlich,3,8,10 Yasmin L. Hurd,1,2,7 and Panos Roussos1,2,3,11 1Department of Psychiatry, 2Friedman Brain Institute, 3Department of Genetics and Genomic Science and Institute for Multiscale Biology, Icahn School of Medicine at Mount Sinai, New York, New York 10029, USA; 4iPSYCH, The Lundbeck Foundation Initiative for Integrative Psychiatric Research, 8000 Aarhus C, Denmark; 5Department of Biomedicine, 6Centre for Integrative Sequencing (iSEQ), Aarhus University, 8000 Aarhus C, Denmark; 7Department of Neuroscience, 8Department of Neurology, Icahn School of Medicine at Mount Sinai, New York, New York 10029, USA; 9Department of Theoretical Computer Science, Faculty of Information Technology, Czech Technical University in Prague, Prague 1600, Czech Republic; 10Department of Pediatrics, Icahn School of Medicine at Mount Sinai, New York, New York 10029, USA; 11Mental Illness Research, Education, and Clinical Center, James J. Peters VA Medical Center, Bronx, New York 10468, USA Most common genetic risk variants associated with neuropsychiatric disease are noncoding and are thought to exert their effects by disrupting the function of cis regulatory elements (CREs), including promoters and enhancers. Within each cell, chromatin is arranged in specific patterns to expose the repertoire of CREs required for optimal spatiotemporal regulation of gene expression. -

The Genetic Overlap Between Mood Disorders and Cardio-Metabolic Diseases: a Systematic
bioRxiv preprint doi: https://doi.org/10.1101/150615; this version posted June 15, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The genetic overlap between mood disorders and cardio-metabolic diseases: A systematic review of genome wide and candidate gene studies Azmeraw T. Amare1*, Klaus Oliver Schubert1, Sarah Cohen-Woods2, Bernhard T. Baune1 *Correspondence to Azmeraw T. Amare [email protected] 1Discipline of Psychiatry, School of Medicine, University of Adelaide, Adelaide, SA, Australia 2School of Psychology, Faculty of Social and Behavioural Sciences, Flinders University, Adelaide, SA, Australia 1 bioRxiv preprint doi: https://doi.org/10.1101/150615; this version posted June 15, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. ABSTRACT Meta-analyses of genome-wide association studies (meta-GWAS) and candidate gene studies have identified genetic variants associated with cardiovascular diseases, metabolic diseases, and mood disorders. Although previous efforts were successful for individual disease conditions (single disease), limited information exists on shared genetic risk between these disorders. This article presents a detailed review and analysis of cardio-metabolic diseases risk (CMD-R) genes that are also associated with mood disorders. Firstly, we reviewed meta-GWA studies published until January 2016, for the diseases "type 2 diabetes, coronary artery disease, hypertension" and/or for the risk factors "blood pressure, obesity, plasma lipid levels, insulin and glucose related traits". We then searched the literature for published associations of these CMD-R genes with mood disorders.