Fishing for Genes in the UCSC Browser a Tutorial
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Analysis of the Calcineurin Pathway Identifies Members of the EGR Gene Family, Specifically EGR3, As Potential Susceptibility Candidates in Schizophrenia
Genetic analysis of the calcineurin pathway identifies members of the EGR gene family, specifically EGR3, as potential susceptibility candidates in schizophrenia Kazuo Yamada*, David J. Gerber†, Yoshimi Iwayama*, Tetsuo Ohnishi*, Hisako Ohba*, Tomoko Toyota*, Jun Aruga‡, Yoshio Minabe*§, Susumu Tonegawa†¶, and Takeo Yoshikawa*ʈ** Laboratories for *Molecular Psychiatry and ‡Comparative Neural Development, RIKEN Brain Science Institute, Saitama 351-0198, Japan; †Howard Hughes Medical Institute and RIKEN–MIT Neuroscience Research Center, The Picower Institute for Learning and Memory, Massachusetts Institute of Technology, Cambridge, MA 02139; §Department of Psychiatry and Neurology, Kanazawa University School of Medicine, Ishikawa 920-8641, Japan; and ʈCore Research for Evolutional Science and Technology, Japan Science and Technology Agency, Saitama 332-0012, Japan Contributed by Susumu Tonegawa, December 7, 2006 (sent for review September 22, 2006) The calcineurin cascade is central to neuronal signal transduction, cineurin is particularly enriched in the CNS, where it plays a critical and genes in this network are intriguing candidate schizophrenia role in the regulation of a diverse array of neuronal functions (5, 6). susceptibility genes. To replicate and extend our previously re- Interestingly, calcineurin is positioned downstream of dopaminer- ported association between the PPP3CC gene, encoding the cal- gic signaling (7) and is involved in NMDA receptor-mediated cineurin catalytic ␥-subunit, and schizophrenia, we examined 84 synaptic plasticity (8) and could therefore provide an important SNPs from 14 calcineurin-related candidate genes for genetic as- functional link between these two neurotransmitter systems. To sociation by using 124 Japanese schizophrenic pedigrees. Four of further explore the involvement of calcineurin dysfunction in these genes (PPP3CC, EGR2, EGR3, and EGR4) showed nominally schizophrenia, we have tested for genetic association of a subset of significant association with schizophrenia. -

Mouse Kcnip2 Conditional Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Kcnip2 Conditional Knockout Project (CRISPR/Cas9) Objective: To create a Kcnip2 conditional knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Kcnip2 gene (NCBI Reference Sequence: NM_145703 ; Ensembl: ENSMUSG00000025221 ) is located on Mouse chromosome 19. 10 exons are identified, with the ATG start codon in exon 1 and the TAG stop codon in exon 10 (Transcript: ENSMUST00000162528). Exon 4 will be selected as conditional knockout region (cKO region). Deletion of this region should result in the loss of function of the Mouse Kcnip2 gene. To engineer the targeting vector, homologous arms and cKO region will be generated by PCR using BAC clone RP23-98F2 as template. Cas9, gRNA and targeting vector will be co-injected into fertilized eggs for cKO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Mice homozygous for disruptions in this gene are susceptible to induced cardiac arrhythmias but are otherwise normal. Exon 4 starts from about 27.65% of the coding region. The knockout of Exon 4 will result in frameshift of the gene. The size of intron 3 for 5'-loxP site insertion: 574 bp, and the size of intron 4 for 3'-loxP site insertion: 532 bp. The size of effective cKO region: ~625 bp. The cKO region does not have any other known gene. Page 1 of 8 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele gRNA region 5' gRNA region 3' 1 2 3 4 5 6 7 8 9 10 Targeting vector Targeted allele Constitutive KO allele (After Cre recombination) Legends Exon of mouse Kcnip2 Homology arm cKO region loxP site Page 2 of 8 https://www.alphaknockout.com Overview of the Dot Plot Window size: 10 bp Forward Reverse Complement Sequence 12 Note: The sequence of homologous arms and cKO region is aligned with itself to determine if there are tandem repeats. -

Egr-1 Induces DARPP-32 Expression in Striatal Medium Spiny Neurons Via a Conserved Intragenic Element
6808 • The Journal of Neuroscience, May 16, 2012 • 32(20):6808–6818 Cellular/Molecular Egr-1 Induces DARPP-32 Expression in Striatal Medium Spiny Neurons via a Conserved Intragenic Element Serene Keilani,1 Samira Chandwani,1 Georgia Dolios,2 Alexey Bogush,3 Heike Beck,4 Antonis K. Hatzopoulos,5 Gadiparthi N. Rao,6 Elizabeth A. Thomas,7 Rong Wang,2 and Michelle E. Ehrlich1,2 Departments of 1Neurology and Pediatrics and 2Genetics and Genomic Sciences, Mount Sinai School of Medicine, New York, New York 10029, 3Weinberg Unit for ALS Research, Department of Neuroscience, Farber Institute for Neuroscience, Thomas Jefferson University, Philadelphia, Pennsylvania 19107, 4Walter Brendel Center of Experimental Medicine, Ludwig Maximilians University, D-81377 Munich, Germany, 5Division of Cardiovascular Medicine, Department of Medicine, Vanderbilt University, Nashville, Tennessee 37240, 6Department of Physiology, University of Tennessee Health Science Center, Memphis, Tennessee 38163, and 7Department of Molecular Biology, The Scripps Research Institute, La Jolla, California 92037 DARPP-32 (dopamine and adenosine 3Ј,5Ј-cyclic monophosphate cAMP-regulated phosphoprotein, 32 kDa) is a striatal-enriched protein that mediates signaling by dopamine and other first messengers in the medium spiny neurons. The transcriptional mechanisms that regulate striatal DARPP-32 expression remain enigmatic and are a subject of much interest in the efforts to induce a striatal phenotype in stem cells. We report the identification and characterization of a conserved region, also known as H10, in intron IV of the gene that codes for DARPP-32 (Ppp1r1b). This DNA sequence forms multiunit complexes with nuclear proteins from adult and embry- onic striata of mice and rats. -

ENCODE Genome-Wide Data on the UCSC Genome Browser Melissa Cline ENCODE Data Coordination Center (DCC) UC Santa Cruz
ENCODE Genome-Wide Data on the UCSC Genome Browser Melissa Cline ENCODE Data Coordination Center (DCC) UC Santa Cruz http://encodeproject.org Slides at http://genome-preview.ucsc.edu/ What is ENCODE? • International consortium project with the goal of cataloguing the functional regions of the human genome GTTTGCCATCTTTTG! CTGCTCTAGGGAATC" CAGCAGCTGTCACCA" TGTAAACAAGCCCAG" GCTAGACCAGTTACC" CTCATCATCTTAGCT" GATAGCCAGCCAGCC" ACCACAGGCATGAGT" • A gold mine of experimental data for independent researchers with available disk space ENCODE covers diverse regulatory processes ENCODE experiments are planned for integrative analysis Example of ENCODE data Genes Dnase HS Chromatin Marks Chromatin State Transcription Factor Binding Transcription RNA Binding Translation ENCODE tracks on the UCSC Genome Browser ENCODE tracks marked with the NHGRI helix There are currently 2061 ENCODE experiments at the ENCODE DCC How to find the data you want Finding ENCODE tracks the hard way A better way to find ENCODE tracks Finding ENCODE metadata descriptions Visualizing: Genome Browser tricks that every ENCODE user should know Turning ENCODE subtracks and views on and off Peaks Signal View on/off Subtrack on/off Right-click to the subtrack display menu Subtrack Drag and Drop Sessions: the easy way to save and share your work Downloading data with less pain 1. Via the Downloads button on the track details page 2. Via the File Selection tool Publishing: the ENCODE data release policy Every ENCODE subtrack has a “Restricted Until” date Key points of the ENCODE data release policy • Anyone is free to download and analyze data. • One cannot submit publications involving ENCODE data unless – the data has been at the ENCODE DCC for at least nine months, or – the data producers have published on the data, or – the data producers have granted permission to publish. -

Psychostimulant-Regulated Plasticity in Interneurons of the Nucleus Accumbens
Psychostimulant-Regulated Plasticity in Interneurons of the Nucleus Accumbens by David A. Gallegos Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Anne E. West, Supervisor ___________________________ Jorg Grandl ___________________________ Debra Silver ___________________________ Gregory Crawford ___________________________ Hiro Matsunami Psychostimulant-Regulated Epigenetic Plasticity in Interneurons of the Nucleus Accumbens submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2019 ABSTRACT Psychostimulant-Regulated Epigenetic Plasticity in Interneurons of the Nucleus Accumbens by David A. Gallegos Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Anne E. West, Supervisor ___________________________ Jorg Grandl ___________________________ Debra Silver ___________________________ Gregory Crawford ___________________________ Hiro Matsunami An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2019 Copyright by David Andres Gallegos 2019 Abstract Exposure to psychostimulant drugs of abuse exerts lasting influences on brain function via the regulation of immediate and persistent gene transcription. These changes in gene transcription drive the development of addictive-like -

DRD2 and PPP1R1B (DARPP-32)
Hettinger et al. Behavioral and Brain Functions 2012, 8:19 http://www.behavioralandbrainfunctions.com/8/1/19 RESEARCH Open Access DRD2 and PPP1R1B (DARPP-32) polymorphisms independently confer increased risk for autism spectrum disorders and additively predict affected status in male-only affected sib-pair families Joe A Hettinger1, Xudong Liu2,3, Melissa L Hudson2,3,4, Alana Lee2,3,4, Ira L Cohen4,5, Ron C Michaelis6, Charles E Schwartz7, Suzanne ME Lewis4,8,9 and Jeanette JA Holden1,2,3,4,10,11* Abstract Background: The neurotransmitter dopamine (DA) modulates executive functions, learning, and emotional processing, all of which are impaired in individuals with autism spectrum disorders (ASDs). Our previous findings suggest a role for dopamine-related genes in families with only affected males. Methods: We examined two additional genes which affect DA function, the DRD2 and PPP1R1B (DARPP-32) genes, in a cohort of 112 male-only affected sib-pair families. Selected polymorphisms spanning these genes were genotyped and both family-based and population-based tests were carried out for association analysis. General discriminant analysis was used to examine the gene-gene interactions in predicting autism susceptibility. Results: There was a significantly increased frequency of the DRD2 rs1800498TT genotype (P = 0.007) in affected males compared to the comparison group, apparently due to over-transmission of the T allele (P = 0.0003). The frequency of the PPP1R1B rs1495099CC genotype in affected males was also higher than that in the comparison group (P = 0.002) due to preferential transmission of the C allele from parents to affected children (P = 0.0009). -

Effects of PPP1R1B (DARPP-32) Polymorphism on Feedback-Related Brain Potentials Across the Life Span
ORIGINAL RESEARCH ARTICLE published: 04 March 2013 doi: 10.3389/fpsyg.2013.00089 Effects of PPP1R1B (DARPP-32) polymorphism on feedback-related brain potentials across the life span Dorothea Hämmerer 1,2*†, Gudio Biele 3,4,Viktor Müller 1, HolgerThiele 5, Peter Nürnberg 5,6,7, Hauke R. Heekeren1,3 and Shu-Chen Li 1,2*† 1 Center for Lifespan Psychology, Max Planck Institute for Human Development, Berlin, Germany 2 Department of Psychology, Technische Universität Dresden, Dresden, Germany 3 Department of Education and Psychology, Freie Universität Berlin, Berlin, Germany 4 Institute of Psychology, University of Oslo, Oslo, Norway 5 Cologne Center for Genomics, University of Cologne, Cologne, Germany 6 Center for Molecular Medicine Cologne, University of Cologne, Cologne, Germany 7 Cologne Excellence Cluster on Cellular Stress Responses in Aging-Associated Diseases, University of Cologne, Cologne, Germany Edited by: Maximizing gains during probabilistic reinforcement learning requires the updating of Alain Dagher, Montreal Neurological choice – outcome expectations at the time when the feedback about a specific choice Institute and Hospital, Canada or action is given. Extant theories and evidence suggest that dopaminergic modulation Reviewed by: Meghana Bhatt, City of Hope, USA plays a crucial role in reinforcement learning and the updating of choice – outcome expec- James F.Cavanagh, Brown University, tations. Furthermore, recently a positive component of the event-related potential about USA 200 ms (P2) after feedback has been suggested to reflect such updating. The efficacy of *Correspondence: dopaminergic modulation changes across the life span. However, to date investigations Dorothea Hämmerer and Shu-Chen of age-related differences in feedback-related P2 during reinforcement learning are still Li, Department of Psychology, Technische Universität Dresden, scarce. -

BIO4342 Exercise 2: Browser-Based Annotation and RNA-Seq Data
BIO4342 Exercise 2: Browser-Based Annotation and RNA-Seq Data Jeremy Buhler March 15, 2010 This exercise continues your introduction to practical issues in comparative annotation. You’ll be annotating genomic sequence from the dot chromosome of Drosophila mojavensis using your knowledge of BLAST and some improved visualization tools. You’ll also consider how best to integrate information from high-throughput sequencing of expressed RNA. 1 Getting Started To begin, go to our local genome browser at http://gander.wustl.edu/. Select “Genome Browser” from the left-side menu and choose the “Improved Dot” assembly of D. mojavensis for viewing. Finally, hit submit to start looking at the sequence. The entire dot assembly is about 1.69 megabases in length; zoom out to see everything. This assembly is built from a set of overlapping fosmid clones prepared for the 2009 edition of BIO 4342. We’ve added a variety of information to the genome browser to help you annotate, such as: • gene-structure predictions from several different tools; • repeats annotated using the RepeatMasker program; • BLAST hits to D. melanogaster proteins; • RNA-Seq data, which we’ll describe in more detail later. Having all this evidence available at once is somewhat overwhelming. To keep the view to a manageable level, I’d suggest that you initially set all the gene prediction tracks (Genscan, Nscan, SNAP, Geneid), as well as the repeat tracks, to “dense” mode, so that each displays on a single line. Set the BLAST hit track (called “D. mel proteins”) to “pack” to see the locations of all BLAST hits, and set the “RNA-Seq Coverage” track to “full” and the “TopHat junctions” track to “pack” to get a detailed view of these results. -

BLAT—The BLAST-Like Alignment Tool
Resource BLAT—The BLAST-Like Alignment Tool W. James Kent Department of Biology and Center for Molecular Biology of RNA, University of California, Santa Cruz, Santa Cruz, California 95064, USA Analyzing vertebrate genomes requires rapid mRNA/DNA and cross-species protein alignments. A new tool, BLAT, is more accurate and 500 times faster than popular existing tools for mRNA/DNA alignments and 50 times faster for protein alignments at sensitivity settings typically used when comparing vertebrate sequences. BLAT’s speed stems from an index of all nonoverlapping K-mers in the genome. This index fits inside the RAM of inexpensive computers, and need only be computed once for each genome assembly. BLAT has several major stages. It uses the index to find regions in the genome likely to be homologous to the query sequence. It performs an alignment between homologous regions. It stitches together these aligned regions (often exons) into larger alignments (typically genes). Finally, BLAT revisits small internal exons possibly missed at the first stage and adjusts large gap boundaries that have canonical splice sites where feasible. This paper describes how BLAT was optimized. Effects on speed and sensitivity are explored for various K-mer sizes, mismatch schemes, and number of required index matches. BLAT is compared with other alignment programs on various test sets and then used in several genome-wide applications. http://genome.ucsc.edu hosts a web-based BLAT server for the human genome. Some might wonder why in the year 2002 the world needs sions on any number of perfect or near-perfect hits. Where another sequence alignment tool. -

No Evidence for Recent Selection at FOXP2 Among Diverse Human Populations
Article No Evidence for Recent Selection at FOXP2 among Diverse Human Populations Graphical Abstract Authors Elizabeth Grace Atkinson, Amanda Jane Audesse, Julia Adela Palacios, Dean Michael Bobo, Ashley Elizabeth Webb, Sohini Ramachandran, Brenna Mariah Henn Correspondence [email protected] (E.G.A.), [email protected] (B.M.H.) In Brief An in-depth examination of diverse sets of human genomes argues against a recent selective evolutionary sweep of FOXP2, a gene that was believed to be critical for speech evolution in early hominins. Highlights d No support for positive selection at FOXP2 in large genomic datasets d Sample composition and genomic scale significantly affect selection scans d An intronic ROI within FOXP2 is expressed in human brain cells and cortical tissue d This ROI contains a large amount of constrained, human- specific polymorphisms Atkinson et al., 2018, Cell 174, 1424–1435 September 6, 2018 ª 2018 Elsevier Inc. https://doi.org/10.1016/j.cell.2018.06.048 Article No Evidence for Recent Selection at FOXP2 among Diverse Human Populations Elizabeth Grace Atkinson,1,8,9,10,* Amanda Jane Audesse,2,3 Julia Adela Palacios,4,5 Dean Michael Bobo,1 Ashley Elizabeth Webb,2,6 Sohini Ramachandran,4 and Brenna Mariah Henn1,7,* 1Department of Ecology and Evolution, Stony Brook University, Stony Brook, NY, USA 2Department of Molecular Biology, Cell Biology and Biochemistry, Brown University, Providence, RI 02912, USA 3Neuroscience Graduate Program, Brown University, Providence, RI 02912, USA 4Department of Ecology and Evolutionary -
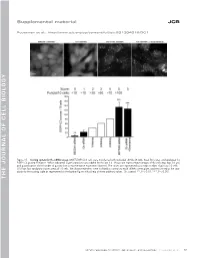
Thejournalofcellb Io Logy
Supplemental material JCB Puustinen et al., http://www.jcb.org/cgi/content/full/jcb.201304012/DC1 Figure S1. Scoring system for the siRNA screen. MCF7-EGFP-LC3 cells were transfected with indicated siRNAs (8 nM), fixed 56 h later, and analyzed for EGFP-LC3 puncta formation. When indicated, 5 µM siramesine was added for the last 3 h. Shown are representative images of the cells (top; bar, 10 µm) and quantification of the number of puncta from a representative experiment (bottom). The values are represented as a mean number of puncta/10 cells ± SDs from four randomly chosen areas of 10 cells. The shown treatments were included as controls to each siRNA screen plate, and they served as the stan- dards for the scoring scale as represented on the bottom figure with a help of three arbitrary values. Ctr, control. **, P < 0.01; ***, P < 0.001. THE JOURNAL OF CELL BIOLOGY CIP2A regulates mTORC1, cell growth, and autophagy • Puustinen et al. S1 Figure S2. A cartoon highlighting the positions of the candidate autophagy-regulating genes in the insulin receptor signaling network. The cartoon was prepared by Ingenuity Pathway Analysis. software. CP, carbohydrate phosphatase; ERK, extracellular signal–regulated kinase; FSH, follicle-stimulating hor- mone; EG, epidermal growth factor. S2 JCB Figure S3. The effect of siRNAs targeting the autophagy-regulating PP2 subunits on target gene expression, cell density, and mTORC1 pathway. (A) Total RNA was isolated from MCF7 cells treated with indicated siRNAs (20 nM) for 54 h and analyzed for target gene expression by qPCR using primers de- signed to specifically amplify the indicated PP2A-related mRNAs andACTB mRNA (internal control). -

A Multithread Blat Algorithm Speeding up Aligning Sequences to Genomes Meng Wang and Lei Kong*
Wang and Kong BMC Bioinformatics (2019) 20:28 https://doi.org/10.1186/s12859-019-2597-8 SOFTWARE Open Access pblat: a multithread blat algorithm speeding up aligning sequences to genomes Meng Wang and Lei Kong* Abstract Background: The blat is a widely used sequence alignment tool. It is especially useful for aligning long sequences and gapped mapping, which cannot be performed properly by other fast sequence mappers designed for short reads. However, the blat tool is single threaded and when used to map whole genome or whole transcriptome sequences to reference genomes this program can take days to finish, making it unsuitable for large scale sequencing projects and iterative analysis. Here, we present pblat (parallel blat), a parallelized blat algorithm with multithread and cluster computing support, which functions to rapidly fine map large scale DNA/RNA sequences against genomes. Results: The pblat algorithm takes advantage of modern multicore processors and significantly reduces the run time with the number of threads used. pblat utilizes almost equal amount of memory as when running blat. The results generated by pblat are identical with those generated by blat. The pblat tool is easy to install and can run on Linux and Mac OS systems. In addition, we provide a cluster version of pblat (pblat-cluster) running on computing clusters with MPI support. Conclusion: pblat is open source and free available for non-commercial users. It is easy to install and easy to use. pblat and pblat-cluster would facilitate the high-throughput mapping of large scale genomic and transcript sequences to reference genomes with both high speed and high precision.