Determining the Diet of New Zealand King Shag Using DNA Metabarcoding
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(Teleostei: Gadiformes: Moridae), Antimora Microlepis and Physiculus Japonicus, from the Western North Pacific
Species Diversity, 2007, 12, 17–27 Pelagic Juveniles of Two Morids (Teleostei: Gadiformes: Moridae), Antimora microlepis and Physiculus japonicus, from the Western North Pacific Makoto Okamoto1, Naoshi Sato2, Takashi Asahida2 and Yoshiro Watanabe3 1 Tohoku National Fisheries Research Institute, 3-27-5 Shinhama-cho, Shiogama, Miyagi, 985-0001 Japan E-mail: [email protected] 2 School of Fisheries Sciences, Kitasato University, 160-4 Okirai, Sanriku-cho, Ofunato, Iwate, 022-0101 Japan 3 Ocean Research Institute, University of Tokyo, Minamidai, Nakano-ku, Tokyo, 164-8639 Japan (Received 1 July 2006; Accepted 20 November 2006) Pelagic juveniles of two morids, Antimora microlepis Bean, 1890 and Physiculus japonicus Hilgendorf, 1879, were collected by midwater trawl (0–20 m depth) from transition waters between the Oyashio and Kuroshio fronts of the western North Pacific in May, 1989. Juveniles of A. microlepis (13 specimens, 30.2–54.3 mm standard length, SL) characteristically have an elongated body, posteriorly positioned anus, 24–25 precaudal vertebrae, chin with a barbel, no ventral luminous organ, elongated pelvic fin rays, and a non-protruding snout. Juveniles of P. japonicus (six specimens, 20.4–39.0 mm SL) characteristically have an elongated body, chin with a barbel, ventral lu- minous organ anterior to the anus, elongated pelvic fin rays, a pointed cau- dal fin, 9–10 dorsal fin rays, 66–70 second dorsal fin rays, 68–72 anal fin rays, six pelvic fin rays, 16ϩ41–42 vertebrae, and ca. 130 longitudinal scales. Key Words: Teleostei, Gadiformes, Moridae, Antimora microlepis, Physicu- lus japonicus, pelagic juvenile. Introduction The family Moridae currently comprises 18 genera and about 110 species, which occur widely from coastal to pelagic waters over the entire world except in the Arctic Ocean (Paulin 1989a; Okamura 1995; Eschmeyer 1998). -

Habitat Use of the Bottlenose Dolphins (Tursiops Truncatus) of Fiordland: Where, Why and the Implications for Management
Habitat use of the bottlenose dolphins (Tursiops truncatus) of Fiordland: Where, why and the implications for management Stephanie M. Bennington A thesis submitted for the degree of Master of Science in Marine Science at the University of Otago, Dunedin, New Zealand. June 2019 Poncho showing off in the beautiful Bowen channel of Dusky Sound photo credit: Steph Bennington i Abstract Understanding the distribution of a species gives important clues about its ecology, and can provide key information and guidance for conservation management. The bottlenose dolphins (Tursiops truncatus) of Fiordland, New Zealand, form three small subpopulations, two of which are mostly resident within separate fjord systems: Doubtful Sound and Dusky Sound. Within these fjords, the dolphins’ distribution and resulting habitat use varies, with high and low use areas, and seasonal variation evident. In this thesis I investigated the distribution patterns of the dolphins, to better understand what drives them and how this relates to the way the dolphins are managed. Specifically, I used Kernel Density Estimation (KDE) from sighting information collected between 2005 and 2018 in Doubtful Sound, and 2009 and 2018 in Dusky Sound, to identify patterns in habitat use over time. Drivers of habitat use were investigated using species distribution models (SDMs), in the form of generalised additive models. Abiotic predictor variables were modelled using long term occurrence data as the response variable. Biotic predictors were included in SDMs for 2018, a year in which I collected data on potential prey and dolphin distribution concurrently. Information on dolphin prey was collected using Baited Underwater Video (BUV). I found that although seasonal variation in habitat use was present, general distribution patterns were consistent through time. -

New Zealand Comprehensive III 16Th November – 2Nd December 2017 Trip Report
New Zealand Comprehensive III 16th November – 2nd December 2017 Trip Report Gibson’s Wandering Albatross off Kaikoura by Erik Forsyth Trip Report compiled by Tour Leader Erik Forsyth Rockjumper Birding Tours | New Zealand www.rockjumperbirding.com Trip Report – RBL New Zealand - Comprehensive III 2017 2 Daily Diary New Zealand is a must for the serious seabird enthusiast. Not only will you see a variety of albatrosses, petrels and shearwaters, but there are good chances of getting out on the high seas and finding something unusual. Seabirds dominate this tour, and views of most birds are right alongside the boat. There are also several land birds which are unique to these islands: the kiwis – terrestrial nocturnal inhabitants; the huge swamp-hen like takahe – prehistoric in its looks and movements; and then the wattlebirds (the saddlebacks and kokako) – poor flyers with short wings, which bound along the branches and on the ground. We had so many highlights on this tour, including close encounters with Little Spotted Kiwi, walk-away views of a pair of North Island Kokako, both North and South Island Saddlebacks and a pair of the impressive South Island Takahe. With many boat trips, the pelagic list was long, with Wandering, Northern and Southern Royal, Salvin’s, Black-browed, Campbell and White-capped Albatrosses, Westland, Cook’s and White- chinned Petrels, Buller’s, Flesh-footed and New Zealand (Red-breasted) Plover by Erik Forsyth Hutton’s Shearwaters, and Common Diving Petrel being a few of the highlights. Other major highlights included twelve New Zealand King Shag, a pair of Blue Duck with one chick, nine of the critically endangered Black Stilt, the rare Stitchbird, New Zealand Kaka, the entertaining Kea, range- restricted New Zealand Rockwren, the tiny Rifleman and lastly the striking Yellowhead, to name a few. -

New Zealand Fishes a Field Guide to Common Species Caught by Bottom, Midwater, and Surface Fishing Cover Photos: Top – Kingfish (Seriola Lalandi), Malcolm Francis
New Zealand fishes A field guide to common species caught by bottom, midwater, and surface fishing Cover photos: Top – Kingfish (Seriola lalandi), Malcolm Francis. Top left – Snapper (Chrysophrys auratus), Malcolm Francis. Centre – Catch of hoki (Macruronus novaezelandiae), Neil Bagley (NIWA). Bottom left – Jack mackerel (Trachurus sp.), Malcolm Francis. Bottom – Orange roughy (Hoplostethus atlanticus), NIWA. New Zealand fishes A field guide to common species caught by bottom, midwater, and surface fishing New Zealand Aquatic Environment and Biodiversity Report No: 208 Prepared for Fisheries New Zealand by P. J. McMillan M. P. Francis G. D. James L. J. Paul P. Marriott E. J. Mackay B. A. Wood D. W. Stevens L. H. Griggs S. J. Baird C. D. Roberts‡ A. L. Stewart‡ C. D. Struthers‡ J. E. Robbins NIWA, Private Bag 14901, Wellington 6241 ‡ Museum of New Zealand Te Papa Tongarewa, PO Box 467, Wellington, 6011Wellington ISSN 1176-9440 (print) ISSN 1179-6480 (online) ISBN 978-1-98-859425-5 (print) ISBN 978-1-98-859426-2 (online) 2019 Disclaimer While every effort was made to ensure the information in this publication is accurate, Fisheries New Zealand does not accept any responsibility or liability for error of fact, omission, interpretation or opinion that may be present, nor for the consequences of any decisions based on this information. Requests for further copies should be directed to: Publications Logistics Officer Ministry for Primary Industries PO Box 2526 WELLINGTON 6140 Email: [email protected] Telephone: 0800 00 83 33 Facsimile: 04-894 0300 This publication is also available on the Ministry for Primary Industries website at http://www.mpi.govt.nz/news-and-resources/publications/ A higher resolution (larger) PDF of this guide is also available by application to: [email protected] Citation: McMillan, P.J.; Francis, M.P.; James, G.D.; Paul, L.J.; Marriott, P.; Mackay, E.; Wood, B.A.; Stevens, D.W.; Griggs, L.H.; Baird, S.J.; Roberts, C.D.; Stewart, A.L.; Struthers, C.D.; Robbins, J.E. -

Biogenic Habitats on New Zealand's Continental Shelf. Part II
Biogenic habitats on New Zealand’s continental shelf. Part II: National field survey and analysis New Zealand Aquatic Environment and Biodiversity Report No. 202 E.G. Jones M.A. Morrison N. Davey S. Mills A. Pallentin S. George M. Kelly I. Tuck ISSN 1179-6480 (online) ISBN 978-1-77665-966-1 (online) September 2018 Requests for further copies should be directed to: Publications Logistics Officer Ministry for Primary Industries PO Box 2526 WELLINGTON 6140 Email: [email protected] Telephone: 0800 00 83 33 Facsimile: 04-894 0300 This publication is also available on the Ministry for Primary Industries websites at: http://www.mpi.govt.nz/news-and-resources/publications http://fs.fish.govt.nz go to Document library/Research reports © Crown Copyright – Fisheries New Zealand TABLE OF CONTENTS EXECUTIVE SUMMARY 1 1. INTRODUCTION 3 1.1 Overview 3 1.2 Objectives 4 2. METHODS 5 2.1 Selection of locations for sampling. 5 2.2 Field survey design and data collection approach 6 2.3 Onboard data collection 7 2.4 Selection of core areas for post-voyage processing. 8 Multibeam data processing 8 DTIS imagery analysis 10 Reference libraries 10 Still image analysis 10 Video analysis 11 Identification of biological samples 11 Sediment analysis 11 Grain-size analysis 11 Total organic matter 12 Calcium carbonate content 12 2.5 Data Analysis of Core Areas 12 Benthic community characterization of core areas 12 Relating benthic community data to environmental variables 13 Fish community analysis from DTIS video counts 14 2.6 Synopsis Section 15 3. RESULTS 17 3.1 -

HOME RANGES and DIVING BEHAVIOR of NEW ZEALAND SEA LIONS ALONG the CATLINS COAST, SOUTH ISLAND, NEW ZEALAND a Dissertation by NA
HOME RANGES AND DIVING BEHAVIOR OF NEW ZEALAND SEA LIONS ALONG THE CATLINS COAST, SOUTH ISLAND, NEW ZEALAND A Dissertation by NATHAN MARK REED Submitted to the Office of Graduate and Professional Studies of Texas A&M University in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Chair of Committee, Randall Davis Committee Members, William Seitz Blair Sterba-Boatwright Bernd Würsig Intercollegiate Faculty Chair, Anja Schulze May 2021 Major Subject: Marine Biology Copyright 2021 Nathan Reed ABSTRACT New Zealand sea lions (Phocarctos hookeri) were extirpated from the North and South Islands of New Zealand during pre-European native hunting, and their numbers were greatly reduced on the Auckland and Campbell Islands during European commercial sealing. However, they began reoccupying South Island in 1994, and pup production remains low but steady. The home range, at-sea movements, and diving behavior of females at the breeding colony along the Catlins Coast of South Island have not been studied since its inception in 2006. The goal of the study was to: 1) evaluate the performance of home range models to identify the most accurate model(s) for a semi- aquatic distribution, 2) track movements to identify home ranges, and 3) record diving behavior of females to characterize foraging behavior and estimate energy expenditure. To accomplish this study, we attached satellite telemeters and video and data recorders to females along the Catlins Coast during austral winter of 2019. Home ranges were most accurately modeled by separating inshore and offshore habitats and applying adaptive local convex hulls (LOCOH) and fixed kernel density with plug-in bandwidth selection (PKDE), respectively. -
2018 Final LOFF W/ Ref and Detailed Info
Final List of Foreign Fisheries Rationale for Classification ** (Presence of mortality or injury (P/A), Co- Occurrence (C/O), Company (if Source of Marine Mammal Analogous Gear Fishery/Gear Number of aquaculture or Product (for Interactions (by group Marine Mammal (A/G), No RFMO or Legal Target Species or Product Type Vessels processor) processing) Area of Operation or species) Bycatch Estimates Information (N/I)) Protection Measures References Detailed Information Antigua and Barbuda Exempt Fisheries http://www.fao.org/fi/oldsite/FCP/en/ATG/body.htm http://www.fao.org/docrep/006/y5402e/y5402e06.htm,ht tp://www.tradeboss.com/default.cgi/action/viewcompan lobster, rock, spiny, demersal fish ies/searchterm/spiny+lobster/searchtermcondition/1/ , (snappers, groupers, grunts, ftp://ftp.fao.org/fi/DOCUMENT/IPOAS/national/Antigua U.S. LoF Caribbean spiny lobster trap/ pot >197 None documented, surgeonfish), flounder pots, traps 74 Lewis Fishing not applicable Antigua & Barbuda EEZ none documented none documented A/G AndBarbuda/NPOA_IUU.pdf Caribbean mixed species trap/pot are category III http://www.nmfs.noaa.gov/pr/interactions/fisheries/tabl lobster, rock, spiny free diving, loops 19 Lewis Fishing not applicable Antigua & Barbuda EEZ none documented none documented A/G e2/Atlantic_GOM_Caribbean_shellfish.html Queen conch (Strombus gigas), Dive (SCUBA & free molluscs diving) 25 not applicable not applicable Antigua & Barbuda EEZ none documented none documented A/G U.S. trade data Southeastern U.S. Atlantic, Gulf of Mexico, and Caribbean snapper- handline, hook and grouper and other reef fish bottom longline/hook-and-line/ >5,000 snapper line 71 Lewis Fishing not applicable Antigua & Barbuda EEZ none documented none documented N/I, A/G U.S. -

Marine Discharge Consent Application ‐ Deck Drainage
MARINE DISCHARGE CONSENT APPLICATION ‐ DECK DRAINAGE Taranaki Basin Prepared for: OMV New Zealand Limited The Majestic Centre Level 20, 100 Willis Street PO Box 2621, Wellington 6015 New Zealand SLR Ref: 740.10078.00000 Version No: ‐v1.0 March 2018 OMV New Zealand Limited SLR Ref No: 740.10078.00000‐R01 Marine Discharge Consent Application ‐ Deck Drainage Filename: 740.10078.00000‐R01‐v1.0 Marine Discharge Consent Taranaki Basin 20180326 (FINAL).docx March 2018 PREPARED BY SLR Consulting NZ Limited Company Number 2443058 5 Duncan Street Port Nelson 7010, Nelson New Zealand T: +64 274 898 628 E: [email protected] www.slrconsulting.com BASIS OF REPORT This report has been prepared by SLR Consulting NZ Limited with all reasonable skill, care and diligence, and taking account of the timescale and resources allocated to it by agreement with OMV New Zealand Limited. Information reported herein is based on the interpretation of data collected, which has been accepted in good faith as being accurate and valid. This report is for the exclusive use of OMV New Zealand Limited. No warranties or guarantees are expressed or should be inferred by any third parties. This report may not be relied upon by other parties without written consent from SLR SLR disclaims any responsibility to the Client and others in respect of any matters outside the agreed scope of the work. DOCUMENT CONTROL Reference Date Prepared Checked Authorised 740.10078.00000‐R01‐v1.0 26 March 2018 SLR Consulting NZ Ltd Dan Govier Dan Govier 740.10078.00000‐R01‐v1.0 Marine Discharge Consent 20180326 (FINAL).docx Page 2 OMV New Zealand Limited SLR Ref No: 740.10078.00000‐R01 Marine Discharge Consent Application ‐ Deck Drainage Filename: 740.10078.00000‐R01‐v1.0 Marine Discharge Consent Taranaki Basin 20180326 (FINAL).docx March 2018 EXECUTIVE SUMMARY OMV New Zealand Limited (OMV New Zealand) is applying for a Marine Discharge Consent (hereafter referred to as a Discharge Consent) under Section 38 of the Exclusive Economic Zone and Continental Shelf (Environmental Effects) Act 2012 (EEZ Act). -

Marlborough Sounds Wildlife Identification Guide
MARLBOROUGH SOUNDS WILDLIFE IDENTIFICATION GUIDE I NZKS operates several Salmon Farms in the Marlborough Sounds. NZKS works continuously to manage and minimise our impact on marine mammals and sea birds within the surrounding environment. An important part of our social license is to monitor wildlife activity around our farms, of which there is an abundance. Reported sightings of a specific species like King Shag can help identify patterns and trends and improve the science-based knowledge around these species and their behaviours. Information captured will also support consent requirements and future consent applications. Mammals and seabirds are instinctively attracted to the farms for shelter and food. Our focus is on minimising the risks in our daily activities and on working in harmony. This document will outline species that could visit our farms. Please use this document to help you identify any wildlife you are unsure of. Please do your utmost NOT to interact with any wild mammal or seabird, but in an event where you need to interact with them please refer to our NZKS Marine Mammal and Shark Management Plan to identify the best course of action. In the event of a wild animal dying in and around our farm, notify your manager as soon as possible. He or she will report it to the Department of Conservation (DoC) and guide you on the next course of action. Please report details of sightings or entanglements of these species to your manager: • ANY stranded/in danger wildlife • Tagged Fur seal • Leopard seal • Southern right whales SEALS • Great white II • Basking sharks • New Zealand King Shag CONTENTS Seals ............................. -
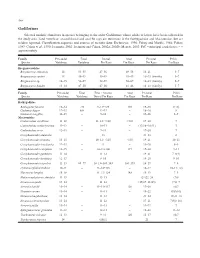
Gadiformes Selected Meristic Characters in Species Belonging to the Order Gadiformes Whose Adults Or Larvae Have Been Collected in the Study Area
548 Gadiformes Selected meristic characters in species belonging to the order Gadiformes whose adults or larvae have been collected in the study area. Total vertebrae, second dorsal and anal fin rays are numerous in the Bathygadidae and Macrouridae, but are seldom reported. Classification sequence and sources of meristic data: Eschmeyer, 1990; Fahay and Markle, 1984; Fahay, 1989; Cohen et al., 1990; Iwamoto, 2002; Iwamoto and Cohen, 2002a; 2002b; Merrett, 2003. PrC = principal caudal rays; ~ = approximately Family Precaudal Total Dorsal Anal Pectoral Pelvic Species Vertebrae Vertebrae Fin Rays Fin Rays Fin Rays Fin Rays Bregmacerotidae Bregmaceros atlanticus 14 53–55 47–56 49–58 16–21 5–7 Bregmaceros cantori 14 45–49 45–49 45–49 16–23 (family) 5–7 Bregmaceros sp. 14–15 52–59 52–59 58–69 16–23 (family) 5–7 Bregmaceros houdei 13–14 47–50 47–50 41–46 16–23 (family) 5–7 Family Precaudal Total First + Second Anal Pectoral Pelvic Species Vertebrae Vertebrae Dorsal Fin Rays Fin Rays Fin Rays Fin Rays Bathygadidae Bathygadus favosus 12–14 ~70 9–11+125 110 15–18 9(10) Gadomus dispar 12–13 80+ 12–13 – 18–20 8 Gadomus longifilis 11–13 – 9–11 – 14–16 8–9 Macrouridae Caelorinchus caribbeus 11–12 – 11–12+>110 >110 17–20 7 Caelorinchus coelorhynchus 11–12 – 10–11 – (17)18–20(21) 7 Caelorinchus occa 12–13 – 9–11 – 17–20 7 Coryphaenoides alateralis – 13 – 21–23 8 Coryphaenoides armatus 13–15 – 10–12+~125 ~135 19–21 10–11 Coryphaenoides brevibarbis 12–13 – 9 – 19–20 8–9 Coryphaenoides carapinus 12–15 – 10–11+100 117 17–20 9–11 Coryphaenoides guentheri -

Rapid Radiation of Southern Ocean Shags in Response to Receding Sea Ice 2 3 Running Title: Blue-Eyed Shag Phylogeography 4 5 Nicolas J
bioRxiv preprint doi: https://doi.org/10.1101/2021.08.18.456742; this version posted August 19, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 1 Rapid radiation of Southern Ocean shags in response to receding sea ice 2 3 Running title: Blue-eyed shag phylogeography 4 5 Nicolas J. Rawlence1, *, Alexander T. Salis1, 2, Hamish G. Spencer1, Jonathan M. Waters1, 6 Lachie Scarsbrook1, Richard A. Phillips3, Luciano Calderón4, Timothée R. Cook5, Charles- 7 André Bost6, Ludovic Dutoit1, Tania M. King1, Juan F. Masello7, Lisa J. Nupen8, Petra 8 Quillfeldt7, Norman Ratcliffe3, Peter G. Ryan5, Charlotte E. Till1, 9, Martyn Kennedy1,* 9 1 Department of Zoology, University of Otago, Dunedin, New Zealand. 10 2 Australian Centre for Ancient DNA, University of Adelaide, South Australia, Australia. 11 3 British Antarctic Survey, Natural Environment Research Council, United Kingdom. 12 4 Instituto de Biología Agrícola de Mendoza (IBAM, CONICET-UNCuyo), Argentina. 13 5 FitzPatrick Institute of African Ornithology, Department of Biological Sciences, University 14 of Cape Town, South Africa. 15 6 CEBC-CNRS, UMR 7372, 405 Route de Prissé la Charrière, 79360 Villiers en Bois, 16 France. 17 7 Justus Liebig University, Giessen, Germany. 18 8 Organisation for Tropical Studies, Skukuza, South Africa. 19 9 School of Human Evolution and Social Change, Arizona State University, Arizona, USA. 20 21 Prepared for submission as a research article to Journal of Biogeography 22 23 * Corresponding authors: [email protected]; [email protected] 24 25 ACKNOWLEDGEMENTS 26 This work was supported with funding from the University of Otago. -

Contrast in the Importance of Arrow Squid As Prey of Male New Zealand Sea Lions and New Zealand Fur Seals at the Snares, Subantarctic New Zealand
Mar Biol (2014) 161:631–643 DOI 10.1007/s00227-013-2366-6 ORIGINAL PAPER Contrast in the importance of arrow squid as prey of male New Zealand sea lions and New Zealand fur seals at The Snares, subantarctic New Zealand Chris Lalas · Trudi Webster Received: 27 April 2013 / Accepted: 3 December 2013 / Published online: 19 December 2013 © Springer-Verlag Berlin Heidelberg 2013 Abstract New Zealand sea lions (Phocarctos hookeri) Introduction are threatened by incidental bycatch in the trawl fishery for southern arrow squid (Nototodarus sloanii). An over- New Zealand sea lions (Phocarctos hookeri), hereafter lap between the fishery and foraging sea lions has previ- referred to as NZ sea lions, have a breeding distribution ously been interpreted as one piece of evidence supporting extending from Otago Peninsula (South Island, New Zea- resource competition for squid. However, there is currently land) south to subantarctic Campbell Island (Fig. 1). The no consensus about the importance of squid in the diet of species is classified as ‘Vulnerable’ by the International New Zealand sea lions. Therefore, we investigated this Union for the Conservation of Nature (IUCN) due to its importance independently of spatial and temporal differ- small population and restricted distribution (IUCN 2012). ences in squid availability through a simultaneous study New Zealand sea lions are the only endemic pinniped spe- with sympatric New Zealand fur seals (Arctocephalus for- cies in New Zealand and 86 % of the entire population steri), a species known to target arrow squid. Diet sampling breed at the subantarctic Auckland Islands (Chilvers et al. at The Snares (48°01′S 166°32′E), subantarctic New Zea- 2007; Chilvers 2008; Fig.