Developmental Therapeutics Immunotherapy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(PDCO) Minutes of the Meeting on 29 January - 01 February 2019
1 March 2019 EMA/PDCO/56017/2019 Inspections, Human Medicines Pharmacovigilance and Committees Division Paediatric Committee (PDCO) Minutes of the meeting on 29 January - 01 February 2019 Chair: Dirk Mentzer – Vice-Chair: Koenraad Norga 29 January 2019, 14:00- 19:00, room 3A 30 January 2019, 08:30- 19:00, room 3A 31 January 2019, 08:30- 19:00, room 3A 01 February 2019, 08:30- 13:00, room 3A Disclaimers Some of the information contained in this set of minutes is considered commercially confidential or sensitive and therefore not disclosed. With regard to intended therapeutic indications or procedure scopes listed against products, it must be noted that these may not reflect the full wording proposed by applicants and may also vary during the course of the review. Additional details on some of these procedures will be published in the PDCO Committee meeting reports (after the PDCO Opinion is adopted), and on the Opinions and decisions on paediatric investigation plans webpage (after the EMA Decision is issued). Of note, this set of minutes is a working document primarily designed for PDCO members and the work the Committee undertakes. Further information with relevant explanatory notes can be found at the end of this document. Note on access to documents Some documents mentioned in the minutes cannot be released at present following a request for access to documents within the framework of Regulation (EC) No 1049/2001 as they are subject to on-going procedures for which a final decision has not yet been adopted. They will become public when adopted or considered public according to the principles stated in the Agency policy on access to documents (EMA/127362/2006). -

Predictive QSAR Tools to Aid in Early Process Development of Monoclonal Antibodies
Predictive QSAR tools to aid in early process development of monoclonal antibodies John Micael Andreas Karlberg Published work submitted to Newcastle University for the degree of Doctor of Philosophy in the School of Engineering November 2019 Abstract Monoclonal antibodies (mAbs) have become one of the fastest growing markets for diagnostic and therapeutic treatments over the last 30 years with a global sales revenue around $89 billion reported in 2017. A popular framework widely used in pharmaceutical industries for designing manufacturing processes for mAbs is Quality by Design (QbD) due to providing a structured and systematic approach in investigation and screening process parameters that might influence the product quality. However, due to the large number of product quality attributes (CQAs) and process parameters that exist in an mAb process platform, extensive investigation is needed to characterise their impact on the product quality which makes the process development costly and time consuming. There is thus an urgent need for methods and tools that can be used for early risk-based selection of critical product properties and process factors to reduce the number of potential factors that have to be investigated, thereby aiding in speeding up the process development and reduce costs. In this study, a framework for predictive model development based on Quantitative Structure- Activity Relationship (QSAR) modelling was developed to link structural features and properties of mAbs to Hydrophobic Interaction Chromatography (HIC) retention times and expressed mAb yield from HEK cells. Model development was based on a structured approach for incremental model refinement and evaluation that aided in increasing model performance until becoming acceptable in accordance to the OECD guidelines for QSAR models. -

Educational Objectives Target Audience Abstract Instructions Services for the Disabled Language/Translation Cme Accreditation Cr
TARGET AUDIENCE PROGRAM REGISTRATION This conference is designed for medical, surgical and radiation Tuition: US$350 Physicians in Practice (prior to June 21, 2004) oncologists, as well as obstetricians, gynecologists and other US$400 Physicians (On/After June 21, 2004 & On-Site) physicians interested in leading edge information on breast US$200 Residents, Fellows, Nurses and Allied Health cancer care and the most promising clinical trials for their breast Professionals cancer or high risk patients. US$130 Accompanying Person to Dinner Reception Tuition is complimentary for Patient Advocates / Cancer Survivors EDUCATIONAL OBJECTIVES Tuition includes, admission to scientific sessions, a course syllabus, continental breakfast, refreshment breaks, a cocktail reception on Upon completion of this conference, participants should be able Thursday, July 22nd, and a dinner event on Friday, July 23rd, to: 2004. Understand the basic mechanisms of the development of breast cancer, its natural history, risk factors, genetic There will be an additional $150 fee for each of the three Surgical component and prognostic indicators; Hands-On Sessions and $100 for the Nursing Seminar. It is required that attendees register for the entire conference in order to register Be up-to-date on today’s diagnostic and treatment for the surgical sessions and/or the luncheons. controversial areas of invasive and non-invasive breast cancer Refunds will be made only if written notice of cancellation is received care; prior to May 21, 2004. A $75 fee is charged for all refunds. After Guide patients to the ongoing clinical trials and develop May 21, 2004, no refunds will be made. In cases where a course novel treatment concepts for future clinical trials; is cancelled due to insufficient registrations, a full tuition refund is made. -

RT+IO Therapy in NSCLC Draft 4 Clinical Cancer Research 1
Author Manuscript Published OnlineFirst on June 26, 2018; DOI: 10.1158/1078-0432.CCR-17-3620 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. RT+IO Therapy in NSCLC Draft 4 Clinical Cancer Research Title: The Integration of Radiotherapy With Immunotherapy for the Treatment of Non-Small Cell Lung Cancer Running title: Radiotherapy and Immunotherapy in Non-Small Cell Lung Cancer Eric C. Ko1, David Raben2, Silvia C. Formenti1 1Department of Radiation Oncology, Weill Cornell Medicine, New York, New York 2Department of Radiation Oncology, University of Colorado Anschutz Medical Campus, Aurora, Colorado Corresponding Author: Silvia C. Formenti, New York-Presbyterian/Weill Cornell Medicine, 525 East 68th Street, N-046, Box 169, New York, NY 10065-4885; Phone: 212-746-3608; Fax: 212-746-8850; E-mail: [email protected]. Confirmed Target Journal: Clinical Cancer Research Journal Specs (Review Article): Word Count (limit 3750 words): 4090 Abstract Word Count (unstructured, limit ≤250 words): 209 Number of References (≤75): 79 Number of Figures/Tables (5): 1 table, 3 figures 1 Downloaded from clincancerres.aacrjournals.org on September 24, 2021. © 2018 American Association for Cancer Research. Author Manuscript Published OnlineFirst on June 26, 2018; DOI: 10.1158/1078-0432.CCR-17-3620 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. RT+IO Therapy in NSCLC Draft 4 Clinical Cancer Research Abstract Five-year survival rates for non-small cell lung cancer (NSCLC) range from 14% to 49% for stage I to stage IIIA disease, and are <5% for stage IIIB/IV disease. -

Celldex Therapeutics Presents Phase 1 Study of Varlilumab and Opdivo® at 2017 ASCO Annual Meeting
June 5, 2017 Celldex Therapeutics Presents Phase 1 Study of Varlilumab and Opdivo® at 2017 ASCO Annual Meeting Early signs of clinical activity without increased toxicity observed HAMPTON, N.J., June 05, 2017 (GLOBE NEWSWIRE) -- Celldex Therapeutics, Inc. (Nasdaq:CLDX) announced today data from the Phase 1 portion of a Phase 1/2 dose escalation and cohort expansion study examining the combination of varlilumab, Celldex's CD27 targeting investigational immune-activating antibody, and Bristol-Myers Squibb's anti-PD-1 immunotherapy Opdivo® (nivolumab). Rachel E. Sanborn, M.D., Co-director of the Providence Thoracic Oncology Program; and Phase I Clinical Trials Program, at the Earle A. Chiles Research Institute, Providence Cancer Center, in Portland, Ore. presented results from the study in an oral presentation entitled, "Clinical Results with Combination of Anti-CD27 Agonist Antibody, Varlilumab, with Anti-PD1 Antibody Nivolumab in Advanced Cancer Patients" at the 2017 American Society of Clinical Oncology (ASCO) Annual Meeting in Chicago. The primary objective of the Phase 1 portion (n=36) of the study was to evaluate the safety and tolerability of the combination. The Phase 2 portion of the study is expected to complete enrollment in early 2018. "Combining PD-1 inhibition with a potent T cell activating agent provides the opportunity to broaden the number of patients that benefit from checkpoint blockade," said Dr. Sanborn. "While early, we have evidence that this combination does not add toxicity, can turn some ‘immune-cold' tumors hot, and may have clinical benefit, including in some patients who are not likely to respond to monotherapy. Further elucidating the role of intermittent versus chronic T cell activation through the comparison of alternate varlilumab dosing regimens is an essential component of the ongoing Phase 2 study and could be important in optimizing the potential of this combination." Key Highlights • The majority of patients enrolled in the study had PD-L1 negative tumor at baseline and presented with Stage IV, heavily- pretreated disease. -
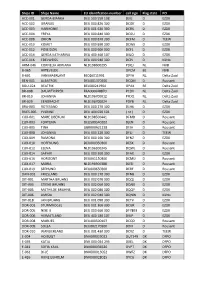
Ships ID Ships Name EU Identifcation Number Call Sign Flag State PO ACC
Ships ID Ships Name EU identifcation number call sign Flag state PO ACC-001 GERDA-BIANKA DEU 500 550 108 DJIG D EZDK ACC-002 URANUS DEU 000 820 300 DCGK D EZDK ACC-003 HARMONIE DEU 001 630 300 DCRK D EZDK ACC-004 FREYA DEU 000 840 300 DCGU D EZDK ACC-008 ORION DEU 000 870 300 DCFM D TEEW ACC-010 KOMET DEU 000 890 300 DCWK D EZDK ACC-012 POSEIDON DEU 000 900 300 DCFL D EZDK ACC-014 GERDA KATHARINA DEU 400 460 107 DIUO D EZDK ACC-016 EDELWEISS DEU 000 940 300 DCPJ D KüNo ARM-046 CORNELIA ADRIANA NLD198600295 PDQJ NL NVB B-065 ARTEVELDE OPCM BE NVB B-601 VAN MAERLANT BE026011991 OPYA NL Delta Zuid BEN-001 ALBATROS DEU001370300 DCQM D Rousant BOU-024 BEATRIX BEL000241964 OPAX BE Delta Zuid BR-008 DAUWTRIPPER FRA000648870 PCDV NL Delta Zuid BR-010 JOHANNA NLD196700012 PFDQ NL Delta Zuid BR-029 EENDRACHT NLD196700024 PDYB NL Delta Zuid BRA-003 ROTESAND DEU 000 270 300 DLHX D EZDK BUES-005 YVONNE DEU 404 020 101 ( nc ) D EZDK CUX-001 MARE LIBERUM NLD198500441 DFMD D Rousant CUX-003 FORTUNA DEU500540102 DJEN D Rousant CUX-005 TINA GBR000A21228 DFJH D Rousant CUX-008 JOHANNA DEU 000 220 300 DFLJ D TEEW CUX-009 RAMONA DEU 000 160 300 DFNZ D EZDK CUX-010 HOFFNUNG DEU000350300 DESX D Rousant CUX-012 ELENA NLD196300345 DQWL D Rousant CUX-014 SAPHIR DEU 000 360 300 DFAX D EZDK CUX-016 HORIZONT DEU001150300 DCMU D Rousant CUX-017 MARIA NLD196900388 DJED D Rousant CUX-019 SEEHUND DEU000650300 DERF D Rousant DAN-001 FRIESLAND DEU 000 170 300 DFNB D EZDK DIT-001 MARTHA BRUHNS DEU 002 070 300 DQQJ D EZDK DIT-003 STIENE BRUHNS DEU 002 060 300 DQNX D EZDK -

LAG-3: from Molecular Functions to Clinical Applications
Open access Review J Immunother Cancer: first published as 10.1136/jitc-2020-001014 on 13 September 2020. Downloaded from LAG-3: from molecular functions to clinical applications Takumi Maruhashi , Daisuke Sugiura , Il- mi Okazaki , Taku Okazaki To cite: Maruhashi T, Sugiura D, ABSTRACT (PD-1) and cytotoxic T lymphocyte antigen Okazaki I, et al. LAG-3: from To prevent the destruction of tissues owing to excessive 4 (CTLA-4) significantly improved the molecular functions to clinical and/or inappropriate immune responses, immune outcomes of patients with diverse cancer applications. Journal for cells are under strict check by various regulatory ImmunoTherapy of Cancer types, revolutionizing cancer treatment. The mechanisms at multiple points. Inhibitory coreceptors, 2020;8:e001014. doi:10.1136/ success of these therapies verified that inhib- including programmed cell death 1 (PD-1) and cytotoxic jitc-2020-001014 itory coreceptors serve as critical checkpoints T lymphocyte antigen 4 (CTLA-4), serve as critical checkpoints in restricting immune responses against for immune cells to not attack the tumor Accepted 29 July 2020 self- tissues and tumor cells. Immune checkpoint inhibitors cells as well as self-tissues. However, response that block PD-1 and CTLA-4 pathways significantly rates are typically lower and immune-related improved the outcomes of patients with diverse cancer adverse events (irAEs) are also observed in types and have revolutionized cancer treatment. However, patients administered with immune check- response rates to such therapies are rather limited, and point inhibitors. This is indicative of the immune-rela ted adverse events are also observed in a continued need to decipher the complex substantial patient population, leading to the urgent need biology of inhibitory coreceptors to increase for novel therapeutics with higher efficacy and lower response rates and prevent such unwanted toxicity. -

Simon Cheng, Victim of a CGTN-Broadcast Forced TV Confession, Files Complaint with UK’S TV Regulator
PRESS RELEASE Simon Cheng, victim of a CGTN-broadcast forced TV confession, files complaint with UK’s TV regulator Madrid, Spain, 2019-11-28: Simon Cheng, Hong Kong resident and former employee at the United Kingdom’s Consulate in Hong Kong, on November 27, 2019 filed an official complaint with Ofcom against CGTN – China Global Television Network - for broadcasting his forced TV confession, extracted while Simon was tortured, held incommunicado, at a secret location, in solitary confinement. The complaint highlights a long list of sections of the UK Broadcasting code that the broadcast violates. “They dragged me into the private van, then instructed me to lay on the rear bench seat. It felt like a kidnapping.” - Simon The complaint by Simon, about a broadcast aired by CGTN on November 21, follows multiple similar complaints earlier. One year ago, UK citizen Peter Humphrey, also with support from Safeguard Defenders, filed a complaint against multiple similar broadcasts of himself and his American wife. Shortly thereafter, Angela Gui, daughter of missing Swedish bookseller Gui Minhai, with support from Safeguard Defenders, filed a complaint against several more broadcasts by CGTN. All these complaints are against broadcasts of forced TV confessions aired before trial, sometimes even before arrest. “I will confess whatever you want, torture is not necessary. They said it is not torture, but ‘training’.” - Simon The broadcast by CGTN comes just one day after Simon Cheng’s explosive interview with BBC and several other news media. The broadcast, which presents accusations as facts, is intended to smear Simon, reduce his credibility, and released almost 3 months after it was actually made, is of little news value. -

9.0 Bn 23 000 200 +
42 | Novartis Annual Report 2017 Innovation The Novartis Institutes for BioMedical Research works in concert with our Global Drug Development group to bring innovative treatments to patients around the world. In 2017, we advanced our drug discovery and development efforts by encouraging greater collaboration and out-of-the-box thinking, exploring new approaches that could improve how we work, and investing in promising tools and technologies. We made progress in priority disease areas with high unmet medical needs. We also marked several key milestones, including US FDA approval – the first of its kind – for a type of personalized cell therapy that could change the course of cancer care. 9.0 bn 23 000 200 + Research and development Scientists, physicians and business Projects in clinical development spending in 2017, amounting to professionals working in research 18.3% of net sales (USD) and development worldwide Collaborative Efficient, effective Progress in important science drug development disease areas We are increasing collaboration We are using digital technology We highlight areas of our work where in research as we try to leverage and data analysis to make drug we are driving significant innovation, innovation from a variety of sources. development swifter and more or where we can potentially have an We are also exploring ways to effective. And we are taking steps important impact on patients and harness digital technology in drug to strengthen our pipeline. public health. discovery, as well as new therapeutic k page 46 k Immuno-oncology page 48 approaches such as cell therapies. k Multiple sclerosis page 50 k page 43 k Liver disease page 52 k Ophthalmology page 53 k Asthma page 55 k Malaria page 56 INNOVATION Novartis Annual Report 2017 | 43 Discovery For new treatments, the journey from laboratory to patient Promoting open innovation starts in the discovery phase where researchers try to Our efforts to increase the flow of ideas between identify potentially groundbreaking therapies. -

China's Practice of Extracting and Broadcasting Forced Conf
Submission: China’s practice of extracting and broadcasting forced confessions before trial ADDITIONAL DATA for Submission to select UN Special Procedures on: China’s practice of extracting and broadcasting forced confessions before trial 2020-08-11 Contact: Benjamin Ismaïl. Safeguard Defenders. [email protected]. +33 663 137 613. Submitted by: Safeguard Defenders ChinaAid Christian Solidarity Worldwide (CSW) Front Line Defenders Human Rights Watch (HRW) Reporters Without Borders (RSF) World Organisation Against Torture (OMCT) 1 Submission: China’s practice of extracting and broadcasting forced confessions before trial (1) OVERVIEW ......................................................................................................................................... 3 (2) Purpose of the present submission .............................................................................................. 4 (3) VIOLATIONS OF NATIONAL AND INTERNATIONAL LAWS ................................................................. 6 (4) Forced confessions: a violation of Chinese laws ........................................................................... 6 (5) Violation of international laws and standards .............................................................................. 8 (6) Right to a fair trial and related rights ........................................................................................ 8 (7) The defects of the Judiciary and International judicial standards ............................................ 9 -

Pembrolizumab Monotherapy in Patients with Previously Treated Metastatic High-Grade Neuroendocrine Neoplasms: Joint Analysis of Two Prospective, Non-Randomised Trials
www.nature.com/bjc ARTICLE Clinical Study Pembrolizumab monotherapy in patients with previously treated metastatic high-grade neuroendocrine neoplasms: joint analysis of two prospective, non-randomised trials Namrata Vijayvergia1, Arvind Dasari2, Mengying Deng1, Samuel Litwin1, Taymeyah Al-Toubah3, R. Katherine Alpaugh1, Efrat Dotan1, Michael J. Hall1, Nicole M. Ross1, Melissa M. Runyen1, Crystal S. Denlinger1, Daniel M. Halperin2, Steven J. Cohen4, Paul F. Engstrom1 and Jonathan R. Strosberg3 BACKGROUND: Metastatic high-grade neuroendocrine neoplasms (G3NENs) have limited treatment options after progression on platinum-based therapy. We addressed the role of Pembrolizumab in patients with previously treated metastatic G3NENs. METHODS: Two open-label, phase 2 studies enrolled patients with G3NEN (Ki-67 > 20%) to receive Pembrolizumab at 200 mg I.V. every 3 weeks. Radiographic evaluation was conducted every 9 weeks with overall response rate as the primary endpoint. RESULTS: Between November 2016 and May 2018, 29 patients (13 males/16 females) with G3NENs were enrolled. One patient (3.4%) had an objective response and an additional six patients (20.7%) had stable disease, resulting in a disease control rate of 24.1%. Disease control rate (DCR) at 18 weeks was 10.3% (3/29). There was no difference in the DCR, PFS or OS between the PD-L1- negative and -positive groups (p 0.56, 0.88 and 0.55, respectively). Pembrolizumab was well tolerated with only 9 grade 3, and no grade 4 events considered drug-related. CONCLUSIONS: Pembrolizumab can be safely administered to patients with G3NENs but has limited activity as a single agent. Successful completion of our trials suggest studies in G3NENs are feasible and present an unmet need. -

The Peoples Republic of China
Wester Hailes High School The Peoples Republic of China Student Name: Class: S3 Modern Studies Teacher: Mr Sinclair- [email protected] Printed & Digital Edition 2021 Modern Studies CfE Level 4 1 Blooms Taxonomy As well as developing your knowledge, this course will also help to equip you with important skills needed to be successful in Modern Studies and the wider world. The success criteria for each lesson will show you the main skills you will use each period. In Modern Studies we aim to promote Higher Order Thinking Skills which encourage a deeper understanding of the information. The following pyramid shows the different levels of thinking skills and as you work your way up the pyramid your learning will become more complex. This should help you to understand the issues covered more thoroughly. Each lesson the aims will be colour coded corresponding to a level on the pyramid so that you know which skills you are using. Modern Studies and the World of Work As a student of Modern Studies you are learning to understand the world around us as well as the political, social and economic issues that affect our lives. The knowledge you gain from your time in Modern Studies will be with you after school and you will refer back to often and in surprising ways. Skills that we practice will prepare you for the future where you will have to create decisions and justify your actions by analysing and evaluating evidence. Our time is known as the “information age” because we are presented with vast amounts of information on an overwhelming level.