NIH Public Access Author Manuscript Curr Allergy Asthma Rep
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

BAFF-Neutralizing Interaction of Belimumab Related to Its Therapeutic Efficacy for Treating Systemic Lupus Erythematosus
ARTICLE DOI: 10.1038/s41467-018-03620-2 OPEN BAFF-neutralizing interaction of belimumab related to its therapeutic efficacy for treating systemic lupus erythematosus Woori Shin1, Hyun Tae Lee1, Heejin Lim1, Sang Hyung Lee1, Ji Young Son1, Jee Un Lee1, Ki-Young Yoo1, Seong Eon Ryu2, Jaejun Rhie1, Ju Yeon Lee1 & Yong-Seok Heo1 1234567890():,; BAFF, a member of the TNF superfamily, has been recognized as a good target for auto- immune diseases. Belimumab, an anti-BAFF monoclonal antibody, was approved by the FDA for use in treating systemic lupus erythematosus. However, the molecular basis of BAFF neutralization by belimumab remains unclear. Here our crystal structure of the BAFF–belimumab Fab complex shows the precise epitope and the BAFF-neutralizing mechanism of belimumab, and demonstrates that the therapeutic activity of belimumab involves not only antagonizing the BAFF–receptor interaction, but also disrupting the for- mation of the more active BAFF 60-mer to favor the induction of the less active BAFF trimer through interaction with the flap region of BAFF. In addition, the belimumab HCDR3 loop mimics the DxL(V/L) motif of BAFF receptors, thereby binding to BAFF in a similar manner as endogenous BAFF receptors. Our data thus provides insights for the design of new drugs targeting BAFF for the treatment of autoimmune diseases. 1 Department of Chemistry, Konkuk University, 120 Neungdong-ro, Gwangjin-gu, Seoul 05029, Republic of Korea. 2 Department of Bio Engineering, Hanyang University, 222 Wangsimni-ro, Seongdong-gu, Seoul 04763, Republic of Korea. These authors contributed equally: Woori Shin, Hyun Tae Lee, Heejin Lim, Sang Hyung Lee. -

Ocrevus (Ocrelizumab) Policy Number: C11250-A
Prior Authorization Criteria Ocrevus (ocrelizumab) Policy Number: C11250-A CRITERIA EFFECTIVE DATES: ORIGINAL EFFECTIVE DATE LAST REVIEWED DATE NEXT REVIEW DUE BY OR BEFORE 8/1/2017 2/17/2021 4/26/2022 LAST P&T J CODE TYPE OF CRITERIA APPROVAL/VERSION J2350-injection,ocrelizumab, Q2 2021 RxPA 1mg 20200428C11250-A PRODUCTS AFFECTED: Ocrevus (ocrelizumab) DRUG CLASS: Multiple Sclerosis Agents - Monoclonal Antibodies ROUTE OF ADMINISTRATION: Intravenous PLACE OF SERVICE: Specialty Pharmacy or Buy and Bill The recommendation is that medications in this policy will be for pharmacy benefit coverage and the IV infusion products administered in a place of service that is a non-hospital facility-based location (i.e., home infusion provider, provider’s office, free-standing ambulatory infusion center) AVAILABLE DOSAGE FORMS: Ocrevus SOLN 300MG/10ML FDA-APPROVED USES: Indicated for the treatment of: • Relapsing forms of multiple sclerosis (MS), to include clinically isolated syndrome, relapsing- remitting disease, and active secondary progressive disease, in adults • Primary progressive MS, in adults COMPENDIAL APPROVED OFF-LABELED USES: None COVERAGE CRITERIA: INITIAL AUTHORIZATION DIAGNOSIS: Multiple Sclerosis REQUIRED MEDICAL INFORMATION: A. RELAPSING FORMS OF MULTIPLE SCLEROSIS: 1. Documentation of a definitive diagnosis of a relapsing form of multiple sclerosis as defined by the McDonald criteria (see Appendix), including: Relapsing- remitting multiple sclerosis [RRMS], secondary-progressive multiple sclerosis [SPMS] with relapses, and progressive- relapsing multiple sclerosis [PRMS] or First clinical episode with MRI features consistent with multiple sclerosis Molina Healthcare, Inc. confidential and proprietary © 2021 This document contains confidential and proprietary information of Molina Healthcare and cannot be reproduced, distributed, or printed without written permission from Molina Healthcare. -

Exploring the Association Between Monoclonal Antibodies and Depression and Suicidal Ideation and Behavior: a Vigibase Study
Drug Safety https://doi.org/10.1007/s40264-018-00789-9 ORIGINAL RESEARCH ARTICLE Exploring the Association between Monoclonal Antibodies and Depression and Suicidal Ideation and Behavior: A VigiBase Study Lotte A. Minnema1,2 · Thijs J. Giezen2,3 · Patrick C. Souverein1 · Toine C. G. Egberts1,4 · Hubert G. M. Leufkens1 · Helga Gardarsdottir1,4,5 © The Author(s) 2019 Abstract Introduction Several monoclonal antibodies (mAbs) have been linked to neuropsychiatric adverse efects in patients, includ- ing depression and suicidal ideation and behavior. Objective The aim of this study was to quantify and characterize spontaneously reported adverse drug reactions (ADRs) of depression and suicidal ideation and behavior related to mAb users, and to explore a possible association with their mecha- nism of action. Methods We included mAb ADRs that were reported in VigiBase, and identifed those related to depression and suicidal ideation and behavior. Reporting odds ratios (RORs) were estimated for each mAb (bevacizumab as the reference) and according to their infuence on the immune system (not directly targeting [reference], stimulating, or suppressing). Those suppressing the immune system were further divided into their intended indication (auto-immune diseases, cancer). Results Overall, 2,924,319 ADRs for 44 mAbs were included; 9455 ADRs were related to depression and 1770 were related to suicidal ideation and behavior. The association was strongest for natalizumab and belimumab, both for depression (ROR 5.7, 95% confdence interval [CI] 5.0–6.4; and ROR 5.1, 95% CI 4.2–6.2) and suicidal ideation and behavior (ROR 12.0, 95% CI 7.9–18.3; and ROR 20.2, 95% CI 12.4–33.0). -

Curriculum Vitae: Daniel J
CURRICULUM VITAE: DANIEL J. WALLACE, M.D., F.A.C.P., M.A.C.R. Up to date as of January 1, 2019 Personal: Address: 8750 Wilshire Blvd, Suite 350 Beverly Hills, CA 90211 Phone: (310) 652-0010 FAX: (310) 360-6219 E mail: [email protected] Education: University of Southern California, 2/67-6/70, BA Medicine, 1971. University of Southern California, 9/70-6/74, M.D, 1974. Postgraduate Training: 7/74-6/75 Medical Intern, Rhode Island (Brown University) Hospital, Providence, RI. 7/75-6/77 Medical Resident, Cedars-Sinai Medical Center, Los Angeles, CA. 7/77-6/79 Rheumatology Fellow, UCLA School of Medicine, Los Angeles, CA. Medical Boards and Licensure: Diplomate, National Board of Medical Examiners, 1975. Board Certified, American Board of Internal Medicine, 1978. Board Certified, Rheumatology subspecialty, 1982. California License: #G-30533. Present Appointments: Medical Director, Wallace Rheumatic Study Center Attune Health Affiliate, Beverly Hills, CA 90211 Attending Physician, Cedars-Sinai Medical Center, Los Angeles, 1979- Clinical Professor of Medicine, David Geffen School of Medicine at UCLA, 1995- Professor of Medicine, Cedars-Sinai Medical Center, 2012- Expert Reviewer, Medical Board of California, 2007- Associate Director, Rheumatology Fellowship Program, Cedars-Sinai Medical Center, 2010- Board of Governors, Cedars-Sinai Medical Center, 2016- Member, Medical Policy Committee, United Rheumatology, 2017- Honorary Appointments: Fellow, American College of Physicians (FACP) Fellow, American College of Rheumatology (FACR) -

Study Protocol
PROTOCOL SYNOPSIS A Multicentre, Randomised, Double-blind, Placebo-controlled, Phase 3 Study Evaluating the Efficacy and Safety of Two Doses of Anifrolumab in Adult Subjects with Active Systemic Lupus Erythematosus International Coordinating Investigator Study site(s) and number of subjects planned Approximately 450 subjects are planned at approximately 173 sites. Study period Phase of development Estimated date of first subject enrolled Q2 2015 3 Estimated date of last subject completed Q2 2018 Study design This is a Phase 3, multicentre, multinational, randomised, double-blind, placebo-controlled study to evaluate the efficacy and safety of an intravenous treatment regimen of anifrolumab (150 mg or 300 mg) versus placebo in subjects with moderately to severely active, autoantibody-positive systemic lupus erythematosus (SLE) while receiving standard of care (SOC) treatment. The study will be performed in adult subjects aged 18 to 70 years of age. Approximately 450 subjects receiving SOC treatment will be randomised in a 1:2:2 ratio to receive a fixed intravenous dose of 150 mg anifrolumab, 300 mg anifrolumab, or placebo every 4 weeks (Q4W) for a total of 13 doses (Week 0 to Week 48), with the primary endpoint evaluated at the Week 52 visit. Investigational product will be administered as an intravenous (IV) infusion via an infusion pump over a minimum of 30 minutes, Q4W. Subjects must be taking either 1 or any combination of the following: oral corticosteroids (OCS), antimalarial, and/or immunosuppressants. Randomisation will be stratified using the following factors: SLE Disease Activity Index 2000 (SLEDAI-2K) score at screening (<10 points versus ≥10 points); Week 0 (Day 1) OCS dose 2(125) Revised Clinical Study Protocol Drug Substance Anifrolumab (MEDI-546) Study Code D3461C00005 Edition Number 5 Date 18 May 2016 (<10 mg/day versus ≥10 mg/day prednisone or equivalent); and results of a type 1 interferon (IFN) test (high versus low). -

Rheumatology and CTD
Section 16 Section Editor: G Narsimulu Rheumatology and CTD 206. Refractory Rheumatoid Arthritis 214. Treatment of SLE beyond Steroids Rohini Handa Rathindra Nath Sarkar, Rudrajit Paul 207. Difficulties in Rheumatoid Arthritis 215. Gout Is the Only Enemy That I Do Not Rajan Kumar Wish to Have at My Feet Anjana Pandey, Ajay Maurya, PK Maheshwari 208. Monoarthritis—A Clinical Dilemma to Internists Arup Kumar Kundu, Abhishek Kundu 216. Liver Dysfunction and NAFLD in RA: 209. Oral Targeted Treatments in RA—Update 2021 Is MTX Really a Culprit? Ramakrishna Rao Uppuluri, Sripurna Deepti Challa Kartik Nikhil Balankhe, Rishabh Ramu Nayak, Pulin Kumar Gupta 210. Biosimilars: Bane or Boon for India 217. Sexual Dysfunction in Rheumatic Diseases Durga Prasanna Misra, Pallavi Patro, Vikas Agarwal Vinod Ravindran 211. An Approach to Vasculitis 218. Interpretation of Common Investigations Packiamary Jerome in Rheumatology 212. How to Manage DMARDs Failure in RA? Renu Saigal, Amit Kansal, Vikram Raj Jain N Subramanian 213. IgG4-related Disease Harpreet Singh, Somdatta Giri, Anju Arya MU-206 (Sec-16).indd 1321 29-01-2021 15:25:29 MU-206 (Sec-16).indd 1322 29-01-2021 15:25:30 CHAPTER 206 Refractory Rheumatoid Arthritis Rohini Handa Abstract A sizeable number of patients with rheumatoid arthritis (RA) are unable to attain low disease activity or remission despite treatment. These difficult to treat (D2T) patients are labeled as refractory RA. The troika of D2T RA, as outlined by the European League against Rheumatism, comprises of treatment failure history, presence of active/symptomatic disease, and clinical perception. The approach to refractory RA is evolving. -

Antagonist Antibodies Against Various Forms of BAFF: Trimer, 60-Mer, and Membrane-Bound S
Supplemental material to this article can be found at: http://jpet.aspetjournals.org/content/suppl/2016/07/19/jpet.116.236075.DC1 1521-0103/359/1/37–44$25.00 http://dx.doi.org/10.1124/jpet.116.236075 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther 359:37–44, October 2016 Copyright ª 2016 by The American Society for Pharmacology and Experimental Therapeutics Unexpected Potency Differences between B-Cell–Activating Factor (BAFF) Antagonist Antibodies against Various Forms of BAFF: Trimer, 60-Mer, and Membrane-Bound s Amy M. Nicoletti, Cynthia Hess Kenny, Ashraf M. Khalil, Qi Pan, Kerry L. M. Ralph, Julie Ritchie, Sathyadevi Venkataramani, David H. Presky, Scott M. DeWire, and Scott R. Brodeur Immune Modulation and Biotherapeutics Discovery, Boehringer Ingelheim Pharmaceuticals, Inc., Ridgefield, Connecticut Received June 20, 2016; accepted July 18, 2016 Downloaded from ABSTRACT Therapeutic agents antagonizing B-cell–activating factor/B- human B-cell proliferation assay and in nuclear factor kB reporter lymphocyte stimulator (BAFF/BLyS) are currently in clinical assay systems in Chinese hamster ovary cells expressing BAFF development for autoimmune diseases; belimumab is the first receptors and transmembrane activator and calcium-modulator Food and Drug Administration–approved drug in more than and cyclophilin ligand interactor (TACI). In contrast to the mouse jpet.aspetjournals.org 50 years for the treatment of lupus. As a member of the tumor system, we find that BAFF trimer activates the human TACI necrosis factor superfamily, BAFF promotes B-cell survival and receptor. Further, we profiled the activities of two clinically ad- homeostasis and is overexpressed in patients with systemic vanced BAFF antagonist antibodies, belimumab and tabalumab. -

1 the BAFF/APRIL System in SLE Pathogenesis. Fabien B Vincent1
. Fabien B Vincent1, Eric F Morand2, Pascal Schneider3 and Fabienne Mackay1 1 Department of Immunology, Monash University, Central Clinical School, Alfred Medical Research and Education Precinct (AMREP), 89 Commercial Road, Melbourne, Victoria 3004, Australia 2 Monash University Centre for Inflammatory Diseases, Southern Clinical School, Monash Medical Centre, 246 Clayton Road, Clayton, Victoria, 3168, Australia 3 Department of Biochemistry, University of Lausanne, Boveresses 155, 1066 Epalinges, Switzerland Abstract Systemic lupus erythematosus (SLE) is characterized by multisystem immune-mediated injury in the setting of autoimmunity to nuclear antigens. The clinical heterogeneity of SLE, the absence of universally agreed clinical trial end points, and the paucity of validated therapeutic targets have, historically, contributed to a lack of novel treatments for SLE. However, in 2011, a therapeutic monoclonal antibody that neutralizes the cytokine TNF ligand superfamily member 13B (also known as B-cell-activating factor of the TNF family [BAFF]), belimumab, became the first targeted therapy for SLE to have efficacy in a randomized clinical trial. Because of its specificity, the efficacy of belimumab provides an opportunity to increase understanding of SLE pathophysiology. Although belimumab depletes B cells, this effect is not as powerful as that of other B-cell-directed therapies that have not been proven efficacious in randomized clinical trials. In this article, therefore, we review results suggesting that neutralizing BAFF can have effects on the immune system other than depletion of B cells. We also identify aspects of the BAFF system for which data in relation to SLE are still missing, and we suggest studies to investigate the pathogenesis of SLE and ways to refine anti-BAFF therapies. -
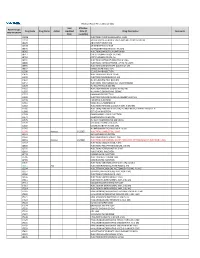
2021 Prior Authorization List Part B Appendix a (PDF)
Medicare Part B PA List Effective 2021 Last Effective Part B Drugs: Drug Code Drug Name Action Updated Date (if Drug Description Comments STEP THERAPY Date available) C9050 INJECTION, EMAPALUMAB-LZSG, 1 MG C9122 MOMETASONE FUROATE SINUS IMPLANT 10 MCG SINUVA J0129 ABATACEPT INJECTION J0178 AFLIBERCEPT INJECTION J0570 BUPRENORPHINE IMPLANT 74.2MG J0585 INJECTION,ONABOTULINUMTOXINA J0717 CERTOLIZUMAB PEGOL INJ 1MG J0718 CERTOLIZUMAB PEGOL INJ J0791 INJECTION CRIZANLIZUMAB-TMCA 5 MG J0800 INJECTION, CORTICOTROPIN, UP TO 40 UNITS J0896 INJECTION LUSPATERCEPT-AAMT 0.25 MG J0897 DENOSUMAB INJECTION J1300 ECULIZUMAB INJECTION J1428 INJECTION ETEPLIRSEN 10 MG J1429 INJECTION GOLODIRSEN 10 MG J1442 INJ FILGRASTIM EXCL BIOSIMIL J1447 INJECTION, TBO-FILGRASTIM, 1 MICROGRAM J1459 INJ IVIG PRIVIGEN 500 MG J1555 INJECTION IMMUNE GLOBULIN 100 MG J1556 INJ, IMM GLOB BIVIGAM, 500MG J1557 GAMMAPLEX INJECTION J1558 INJECTION IMMUNE GLOBULIN XEMBIFY 100 MG J1559 HIZENTRA INJECTION J1561 GAMUNEX-C/GAMMAKED J1562 INJECTION; IMMUNE GLOBULIN 10%, 5 GRAMS J1566 INJECTION, IMMUNE GLOBULIN, INTRAVENOUS, LYOPHILIZED (E.G. P J1568 OCTAGAM INJECTION J1569 GAMMAGARD LIQUID INJECTION J1572 FLEBOGAMMA INJECTION J1575 INJ IG/HYALURONIDASE 100 MG IG J1599 IVIG NON-LYOPHILIZED, NOS J1602 GOLIMUMAB FOR IV USE 1MG J1745 INJ INFLIXIMAB EXCL BIOSIMILR 10 MG J1930 Remove 1/1/2021 INJECTION, LANREOTIDE, 1 MG J2323 NATALIZUMAB INJECTION J2350 INJECTION OCRELIZUMAB 1 MG J2353 Remove 1/1/2021 INJECTION, OCTREOTIDE, DEPOT FORM FOR INTRAMUSCULAR INJECTION, 1 MG J2357 INJECTION, OMALIZUMAB, -

The Future of B-Cell Activating Factor Antagonists in the Treatment of Systemic Lupus Erythematosus
pISSN: 2093-940X, eISSN: 2233-4718 Journal of Rheumatic Diseases Vol. 24, No. 2, April, 2017 https://doi.org/10.4078/jrd.2017.24.2.65 Review Article The Future of B-cell Activating Factor Antagonists in the Treatment of Systemic Lupus Erythematosus William Stohl Division of Rheumatology, Department of Medicine, University of Southern California Keck School of Medicine, Los Angeles, CA, USA To review B-cell activating factor (BAFF)-antagonist therapy in systemic lupus erythematosus (SLE), literature was searched us- ing the search words and phrases, “BAFF”, “B lymphocyte stimulator (BLyS)”, “a proliferation-inducing ligand (APRIL)”, “B-cell maturation antigen (BCMA)”, “transmembrane activator and calcium-modulating and cyclophilin ligand interactor (TACI)”, “BLyS receptor 3 (BR3)”, “belimumab”, “atacicept”, “blisibimod”, “tabalumab”, and “lupus clinical trial”. In addition, papers from the author’s personal library were searched. BAFF-antagonist therapy in SLE has a checkered past, with four late-stage clin- ical trials meeting their primary endpoints and four failing to do so. Additional late-stage clinical trials are enrolling subjects to address some of the remaining unresolved questions, and novel approaches are proposed to improve results. The BAFF-centric pathway is a proven therapeutic target in SLE. As the only pathway in the past 50+ years to have yielded an United States Food and Drug Administration-approved drug for SLE, it occupies a unique place in the armamentarium of the practicing rheumatologist. The challenges facing clinicians and investigators are how to better tweak the BAFF-centric pathway and im- prove on the successes realized. (J Rheum Dis 2017;24:65-73) Key Words. -

Therapeutics Advisory Group
Therapeutics Advisory Group CCG and NHS Trusts in Norfolk and Waveney Index of TAG recommendations Generic name Indication BNFclass Trafficlight IQoro euromuscular training Hiatus hernia - improving symptoms No BNF entry - device Double Red Not recommended for device routine use / Not commissioned (L-) Carnitine Carnitine Deficiency 9.8.1 Drugs used in Red Hospital / Specialist metabolic disorders only (Para-)aminosalicylic acid Tuberculosis 5.1.9 Antituberculosis Double Red Not recommended for drugs - routine use / Not Antimycobacterials commissioned 5-fluorouracil + salicyclic acid Hyperkeratotic actinic keratosis 13.8.1 Photodamage Double-GreenSuitable for GPs to topical solution initiate and prescribe 5-fluorouracil 5% w/w cream Non-hypertrophic actinic keratosis 13.8.1 Photodamage Double-GreenSuitable for GPs to initiate and prescribe Abacavir HIV infection in combination with other 5.3.1 HIV Infection Red Hospital / Specialist antiretroviral drugs only Abacavir + dolutegravir + HIV infection in combination with other 5.3.1 HIV Infection Red Hospital / Specialist lamivudine antiretroviral drugs only Abacavir and lamivudine HIV infection in combination with other 5.3.1 HIV Infection Red Hospital / Specialist antiretroviral drugs only Abaloparatide Male and juvenile osteoporosis 6.6.1 Calcitonin and Double Red Not recommended for Calcitonin and routine use / Not parathyroid hormone commissioned Abatacept Rheumatoid arthritis - 1st line biologic 10.1.3 Drugs that suppress Red Hospital / Specialist after failure of non-biologic DMARDs -

CDER Breakthrough Therapy Designation Approvals Data As of December 31, 2020 Total of 190 Approvals
CDER Breakthrough Therapy Designation Approvals Data as of December 31, 2020 Total of 190 Approvals Submission Application Type and Proprietary Approval Use Number Number Name Established Name Applicant Date Treatment of patients with previously BLA 125486 ORIGINAL-1 GAZYVA OBINUTUZUMAB GENENTECH INC 01-Nov-2013 untreated chronic lymphocytic leukemia in combination with chlorambucil Treatment of patients with mantle cell NDA 205552 ORIGINAL-1 IMBRUVICA IBRUTINIB PHARMACYCLICS LLC 13-Nov-2013 lymphoma (MCL) Treatment of chronic hepatitis C NDA 204671 ORIGINAL-1 SOVALDI SOFOSBUVIR GILEAD SCIENCES INC 06-Dec-2013 infection Treatment of cystic fibrosis patients age VERTEX PHARMACEUTICALS NDA 203188 SUPPLEMENT-4 KALYDECO IVACAFTOR 21-Feb-2014 6 years and older who have mutations INC in the CFTR gene Treatment of previously untreated NOVARTIS patients with chronic lymphocytic BLA 125326 SUPPLEMENT-60 ARZERRA OFATUMUMAB PHARMACEUTICALS 17-Apr-2014 leukemia (CLL) for whom fludarabine- CORPORATION based therapy is considered inappropriate Treatment of patients with anaplastic NOVARTIS lymphoma kinase (ALK)-positive NDA 205755 ORIGINAL-1 ZYKADIA CERITINIB 29-Apr-2014 PHARMACEUTICALS CORP metastatic non-small cell lung cancer (NSCLC) who have progressed on or are intolerant to crizotinib Treatment of relapsed chronic lymphocytic leukemia (CLL), in combination with rituximab, in patients NDA 206545 ORIGINAL-1 ZYDELIG IDELALISIB GILEAD SCIENCES INC 23-Jul-2014 for whom rituximab alone would be considered appropriate therapy due to other co-morbidities