Study Protocol
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

BAFF-Neutralizing Interaction of Belimumab Related to Its Therapeutic Efficacy for Treating Systemic Lupus Erythematosus
ARTICLE DOI: 10.1038/s41467-018-03620-2 OPEN BAFF-neutralizing interaction of belimumab related to its therapeutic efficacy for treating systemic lupus erythematosus Woori Shin1, Hyun Tae Lee1, Heejin Lim1, Sang Hyung Lee1, Ji Young Son1, Jee Un Lee1, Ki-Young Yoo1, Seong Eon Ryu2, Jaejun Rhie1, Ju Yeon Lee1 & Yong-Seok Heo1 1234567890():,; BAFF, a member of the TNF superfamily, has been recognized as a good target for auto- immune diseases. Belimumab, an anti-BAFF monoclonal antibody, was approved by the FDA for use in treating systemic lupus erythematosus. However, the molecular basis of BAFF neutralization by belimumab remains unclear. Here our crystal structure of the BAFF–belimumab Fab complex shows the precise epitope and the BAFF-neutralizing mechanism of belimumab, and demonstrates that the therapeutic activity of belimumab involves not only antagonizing the BAFF–receptor interaction, but also disrupting the for- mation of the more active BAFF 60-mer to favor the induction of the less active BAFF trimer through interaction with the flap region of BAFF. In addition, the belimumab HCDR3 loop mimics the DxL(V/L) motif of BAFF receptors, thereby binding to BAFF in a similar manner as endogenous BAFF receptors. Our data thus provides insights for the design of new drugs targeting BAFF for the treatment of autoimmune diseases. 1 Department of Chemistry, Konkuk University, 120 Neungdong-ro, Gwangjin-gu, Seoul 05029, Republic of Korea. 2 Department of Bio Engineering, Hanyang University, 222 Wangsimni-ro, Seongdong-gu, Seoul 04763, Republic of Korea. These authors contributed equally: Woori Shin, Hyun Tae Lee, Heejin Lim, Sang Hyung Lee. -

Ocrevus (Ocrelizumab) Policy Number: C11250-A
Prior Authorization Criteria Ocrevus (ocrelizumab) Policy Number: C11250-A CRITERIA EFFECTIVE DATES: ORIGINAL EFFECTIVE DATE LAST REVIEWED DATE NEXT REVIEW DUE BY OR BEFORE 8/1/2017 2/17/2021 4/26/2022 LAST P&T J CODE TYPE OF CRITERIA APPROVAL/VERSION J2350-injection,ocrelizumab, Q2 2021 RxPA 1mg 20200428C11250-A PRODUCTS AFFECTED: Ocrevus (ocrelizumab) DRUG CLASS: Multiple Sclerosis Agents - Monoclonal Antibodies ROUTE OF ADMINISTRATION: Intravenous PLACE OF SERVICE: Specialty Pharmacy or Buy and Bill The recommendation is that medications in this policy will be for pharmacy benefit coverage and the IV infusion products administered in a place of service that is a non-hospital facility-based location (i.e., home infusion provider, provider’s office, free-standing ambulatory infusion center) AVAILABLE DOSAGE FORMS: Ocrevus SOLN 300MG/10ML FDA-APPROVED USES: Indicated for the treatment of: • Relapsing forms of multiple sclerosis (MS), to include clinically isolated syndrome, relapsing- remitting disease, and active secondary progressive disease, in adults • Primary progressive MS, in adults COMPENDIAL APPROVED OFF-LABELED USES: None COVERAGE CRITERIA: INITIAL AUTHORIZATION DIAGNOSIS: Multiple Sclerosis REQUIRED MEDICAL INFORMATION: A. RELAPSING FORMS OF MULTIPLE SCLEROSIS: 1. Documentation of a definitive diagnosis of a relapsing form of multiple sclerosis as defined by the McDonald criteria (see Appendix), including: Relapsing- remitting multiple sclerosis [RRMS], secondary-progressive multiple sclerosis [SPMS] with relapses, and progressive- relapsing multiple sclerosis [PRMS] or First clinical episode with MRI features consistent with multiple sclerosis Molina Healthcare, Inc. confidential and proprietary © 2021 This document contains confidential and proprietary information of Molina Healthcare and cannot be reproduced, distributed, or printed without written permission from Molina Healthcare. -

Exploring the Association Between Monoclonal Antibodies and Depression and Suicidal Ideation and Behavior: a Vigibase Study
Drug Safety https://doi.org/10.1007/s40264-018-00789-9 ORIGINAL RESEARCH ARTICLE Exploring the Association between Monoclonal Antibodies and Depression and Suicidal Ideation and Behavior: A VigiBase Study Lotte A. Minnema1,2 · Thijs J. Giezen2,3 · Patrick C. Souverein1 · Toine C. G. Egberts1,4 · Hubert G. M. Leufkens1 · Helga Gardarsdottir1,4,5 © The Author(s) 2019 Abstract Introduction Several monoclonal antibodies (mAbs) have been linked to neuropsychiatric adverse efects in patients, includ- ing depression and suicidal ideation and behavior. Objective The aim of this study was to quantify and characterize spontaneously reported adverse drug reactions (ADRs) of depression and suicidal ideation and behavior related to mAb users, and to explore a possible association with their mecha- nism of action. Methods We included mAb ADRs that were reported in VigiBase, and identifed those related to depression and suicidal ideation and behavior. Reporting odds ratios (RORs) were estimated for each mAb (bevacizumab as the reference) and according to their infuence on the immune system (not directly targeting [reference], stimulating, or suppressing). Those suppressing the immune system were further divided into their intended indication (auto-immune diseases, cancer). Results Overall, 2,924,319 ADRs for 44 mAbs were included; 9455 ADRs were related to depression and 1770 were related to suicidal ideation and behavior. The association was strongest for natalizumab and belimumab, both for depression (ROR 5.7, 95% confdence interval [CI] 5.0–6.4; and ROR 5.1, 95% CI 4.2–6.2) and suicidal ideation and behavior (ROR 12.0, 95% CI 7.9–18.3; and ROR 20.2, 95% CI 12.4–33.0). -

Curriculum Vitae: Daniel J
CURRICULUM VITAE: DANIEL J. WALLACE, M.D., F.A.C.P., M.A.C.R. Up to date as of January 1, 2019 Personal: Address: 8750 Wilshire Blvd, Suite 350 Beverly Hills, CA 90211 Phone: (310) 652-0010 FAX: (310) 360-6219 E mail: [email protected] Education: University of Southern California, 2/67-6/70, BA Medicine, 1971. University of Southern California, 9/70-6/74, M.D, 1974. Postgraduate Training: 7/74-6/75 Medical Intern, Rhode Island (Brown University) Hospital, Providence, RI. 7/75-6/77 Medical Resident, Cedars-Sinai Medical Center, Los Angeles, CA. 7/77-6/79 Rheumatology Fellow, UCLA School of Medicine, Los Angeles, CA. Medical Boards and Licensure: Diplomate, National Board of Medical Examiners, 1975. Board Certified, American Board of Internal Medicine, 1978. Board Certified, Rheumatology subspecialty, 1982. California License: #G-30533. Present Appointments: Medical Director, Wallace Rheumatic Study Center Attune Health Affiliate, Beverly Hills, CA 90211 Attending Physician, Cedars-Sinai Medical Center, Los Angeles, 1979- Clinical Professor of Medicine, David Geffen School of Medicine at UCLA, 1995- Professor of Medicine, Cedars-Sinai Medical Center, 2012- Expert Reviewer, Medical Board of California, 2007- Associate Director, Rheumatology Fellowship Program, Cedars-Sinai Medical Center, 2010- Board of Governors, Cedars-Sinai Medical Center, 2016- Member, Medical Policy Committee, United Rheumatology, 2017- Honorary Appointments: Fellow, American College of Physicians (FACP) Fellow, American College of Rheumatology (FACR) -
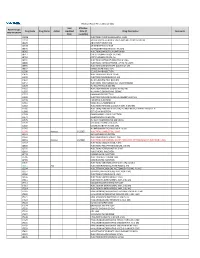
2021 Prior Authorization List Part B Appendix a (PDF)
Medicare Part B PA List Effective 2021 Last Effective Part B Drugs: Drug Code Drug Name Action Updated Date (if Drug Description Comments STEP THERAPY Date available) C9050 INJECTION, EMAPALUMAB-LZSG, 1 MG C9122 MOMETASONE FUROATE SINUS IMPLANT 10 MCG SINUVA J0129 ABATACEPT INJECTION J0178 AFLIBERCEPT INJECTION J0570 BUPRENORPHINE IMPLANT 74.2MG J0585 INJECTION,ONABOTULINUMTOXINA J0717 CERTOLIZUMAB PEGOL INJ 1MG J0718 CERTOLIZUMAB PEGOL INJ J0791 INJECTION CRIZANLIZUMAB-TMCA 5 MG J0800 INJECTION, CORTICOTROPIN, UP TO 40 UNITS J0896 INJECTION LUSPATERCEPT-AAMT 0.25 MG J0897 DENOSUMAB INJECTION J1300 ECULIZUMAB INJECTION J1428 INJECTION ETEPLIRSEN 10 MG J1429 INJECTION GOLODIRSEN 10 MG J1442 INJ FILGRASTIM EXCL BIOSIMIL J1447 INJECTION, TBO-FILGRASTIM, 1 MICROGRAM J1459 INJ IVIG PRIVIGEN 500 MG J1555 INJECTION IMMUNE GLOBULIN 100 MG J1556 INJ, IMM GLOB BIVIGAM, 500MG J1557 GAMMAPLEX INJECTION J1558 INJECTION IMMUNE GLOBULIN XEMBIFY 100 MG J1559 HIZENTRA INJECTION J1561 GAMUNEX-C/GAMMAKED J1562 INJECTION; IMMUNE GLOBULIN 10%, 5 GRAMS J1566 INJECTION, IMMUNE GLOBULIN, INTRAVENOUS, LYOPHILIZED (E.G. P J1568 OCTAGAM INJECTION J1569 GAMMAGARD LIQUID INJECTION J1572 FLEBOGAMMA INJECTION J1575 INJ IG/HYALURONIDASE 100 MG IG J1599 IVIG NON-LYOPHILIZED, NOS J1602 GOLIMUMAB FOR IV USE 1MG J1745 INJ INFLIXIMAB EXCL BIOSIMILR 10 MG J1930 Remove 1/1/2021 INJECTION, LANREOTIDE, 1 MG J2323 NATALIZUMAB INJECTION J2350 INJECTION OCRELIZUMAB 1 MG J2353 Remove 1/1/2021 INJECTION, OCTREOTIDE, DEPOT FORM FOR INTRAMUSCULAR INJECTION, 1 MG J2357 INJECTION, OMALIZUMAB, -

CDER Breakthrough Therapy Designation Approvals Data As of December 31, 2020 Total of 190 Approvals
CDER Breakthrough Therapy Designation Approvals Data as of December 31, 2020 Total of 190 Approvals Submission Application Type and Proprietary Approval Use Number Number Name Established Name Applicant Date Treatment of patients with previously BLA 125486 ORIGINAL-1 GAZYVA OBINUTUZUMAB GENENTECH INC 01-Nov-2013 untreated chronic lymphocytic leukemia in combination with chlorambucil Treatment of patients with mantle cell NDA 205552 ORIGINAL-1 IMBRUVICA IBRUTINIB PHARMACYCLICS LLC 13-Nov-2013 lymphoma (MCL) Treatment of chronic hepatitis C NDA 204671 ORIGINAL-1 SOVALDI SOFOSBUVIR GILEAD SCIENCES INC 06-Dec-2013 infection Treatment of cystic fibrosis patients age VERTEX PHARMACEUTICALS NDA 203188 SUPPLEMENT-4 KALYDECO IVACAFTOR 21-Feb-2014 6 years and older who have mutations INC in the CFTR gene Treatment of previously untreated NOVARTIS patients with chronic lymphocytic BLA 125326 SUPPLEMENT-60 ARZERRA OFATUMUMAB PHARMACEUTICALS 17-Apr-2014 leukemia (CLL) for whom fludarabine- CORPORATION based therapy is considered inappropriate Treatment of patients with anaplastic NOVARTIS lymphoma kinase (ALK)-positive NDA 205755 ORIGINAL-1 ZYKADIA CERITINIB 29-Apr-2014 PHARMACEUTICALS CORP metastatic non-small cell lung cancer (NSCLC) who have progressed on or are intolerant to crizotinib Treatment of relapsed chronic lymphocytic leukemia (CLL), in combination with rituximab, in patients NDA 206545 ORIGINAL-1 ZYDELIG IDELALISIB GILEAD SCIENCES INC 23-Jul-2014 for whom rituximab alone would be considered appropriate therapy due to other co-morbidities -

SUPPLEMENTARY APPENDIX 4: Search Strategies Syntax Guide For
SUPPLEMENTARY APPENDIX 4: Search Strategies Pubmed, Embase Perioperative Management - PubMed Search Strategy – March 6, 2016 Syntax Guide for PubMed [MH] = Medical Subject Heading, also [TW] = Includes all words and numbers in known as MeSH the title, abstract, other abstract, MeSH terms, MeSH Subheadings, Publication Types, Substance Names, Personal Name as Subject, Corporate Author, Secondary Source, Comment/Correction Notes, and Other Terms - typically non-MeSH subject terms (keywords)…assigned by an organization other than NLM [SH] = a Medical Subject Heading [TIAB] = Includes words in the title and subheading, e.g. drug therapy abstracts [MH:NOEXP] = a command to retrieve the results of the Medical Subject Heading specified, but not narrower Medical Subject Heading terms Boolean Operators OR = retrieves results that include at least AND = retrieves results that include all the one of the search terms search terms NOT = excludes the retrieval of terms from the search Perioperative Management PubMed Search Strategy – March 6, 2016 Search Query #1 ((((ARTHROPLASTY, REPLACEMENT, HIP[MH] OR HIP PROSTHES*[TW] OR HIP REPLACEMENT*[TIAB] OR HIP ARTHROPLAST*[TIAB] OR HIP TOTAL REPLACEMENT*[TIAB] OR FEMORAL HEAD PROSTHES*[TIAB]) OR (ARTHROPLASTY, REPLACEMENT, KNEE[MH] OR KNEE PROSTHES*[TW] OR KNEE REPLACEMENT*[TW] OR KNEE ARTHROPLAST*[TW] OR KNEE TOTAL REPLACEMENT*[TIAB]) OR (ARTHROPLAST*[TW] AND (HIP[TIAB] OR HIPS[TIAB] OR KNEE*[TIAB])) AND (("1980/01/01"[PDAT] : "2016/03/06"[PDAT]) AND ENGLISH[LANG])) NOT (((("ADOLESCENT"[MESH]) OR "CHILD"[MESH]) -

(12) Patent Application Publication (10) Pub. No.: US 2017/0172932 A1 Peyman (43) Pub
US 20170172932A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2017/0172932 A1 Peyman (43) Pub. Date: Jun. 22, 2017 (54) EARLY CANCER DETECTION AND A 6LX 39/395 (2006.01) ENHANCED IMMUNOTHERAPY A61R 4I/00 (2006.01) (52) U.S. Cl. (71) Applicant: Gholam A. Peyman, Sun City, AZ CPC .......... A61K 9/50 (2013.01); A61K 39/39558 (US) (2013.01); A61K 4I/0052 (2013.01); A61 K 48/00 (2013.01); A61K 35/17 (2013.01); A61 K (72) Inventor: sham A. Peyman, Sun City, AZ 35/15 (2013.01); A61K 2035/124 (2013.01) (21) Appl. No.: 15/143,981 (57) ABSTRACT (22) Filed: May 2, 2016 A method of therapy for a tumor or other pathology by administering a combination of thermotherapy and immu Related U.S. Application Data notherapy optionally combined with gene delivery. The combination therapy beneficially treats the tumor and pre (63) Continuation-in-part of application No. 14/976,321, vents tumor recurrence, either locally or at a different site, by filed on Dec. 21, 2015. boosting the patient’s immune response both at the time or original therapy and/or for later therapy. With respect to Publication Classification gene delivery, the inventive method may be used in cancer (51) Int. Cl. therapy, but is not limited to such use; it will be appreciated A 6LX 9/50 (2006.01) that the inventive method may be used for gene delivery in A6 IK 35/5 (2006.01) general. The controlled and precise application of thermal A6 IK 4.8/00 (2006.01) energy enhances gene transfer to any cell, whether the cell A 6LX 35/7 (2006.01) is a neoplastic cell, a pre-neoplastic cell, or a normal cell. -

Tysabri® (Natalizumab)
UnitedHealthcare® Commercial Medical Benefit Drug Policy Tysabri® (Natalizumab) Policy Number: 2021D0026M Effective Date: May 1, 2021 Instructions for Use Table of Contents Page Community Plan Policy Coverage Rationale ....................................................................... 1 • Tysabri® (Natalizumab) Applicable Codes .......................................................................... 2 Background.................................................................................... 3 Benefit Considerations .................................................................. 3 Clinical Evidence ........................................................................... 4 U.S. Food and Drug Administration ............................................. 6 References ..................................................................................... 7 Policy History/Revision Information ............................................. 8 Instructions for Use ....................................................................... 8 Coverage Rationale See Benefit Considerations Tysabri (natalizumab) is proven for the treatment of: Relapsing Forms of Multiple Sclerosis Tysabri (natalizumab) is medically necessary for the treatment of relapsing forms of multiple sclerosis (MS) when all of the following are met:1 Initial Therapy o Diagnosis of relapsing forms of multiple sclerosis (MS) (e.g., clinically isolated syndrome, relapsing-remitting disease, active secondary-progressive disease); and o Patient is not receiving Tysabri -

NIH Public Access Author Manuscript Curr Allergy Asthma Rep
NIH Public Access Author Manuscript Curr Allergy Asthma Rep. Author manuscript; available in PMC 2013 December 01. Published in final edited form as: Curr Allergy Asthma Rep. 2012 December ; 12(6): 495–510. doi:10.1007/s11882-012-0307-y. Cellular Targeting in Autoimmunity Jennifer L. Rogers, Donald S. Serafin, Roman G. Timoshchenko, and Teresa K. Tarrant $watermark-textUniversity $watermark-text of North $watermark-text Carolina School of Medicine Division of Rheumatology, Allergy, and Immunology and the Thurston Arthritis Research Center CB#7280, 3300 Manning Dr. Chapel Hill, NC 27517 Abstract Many biologic agents that were first approved for the treatment of malignancies are now being actively investigated and used in a variety of autoimmune diseases such as rheumatoid arthritis (RA), antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis, systemic lupus erythematosus (SLE), and Sjogren’s syndrome. The relatively recent advance of selective immune targeting has significantly changed the management of autoimmune disorders, and in part, can be attributed to the progress made in understanding effector cell function and their signaling pathways. In this review, we will discuss the recent FDA approved biologic therapies that directly target immune cells as well as the most promising investigational drugs affecting immune cell function and signaling for the treatment of autoimmune disease. Keywords Autoimmune disease; Autoimmunity; Biologic therapy; Rheumatoid arthritis; Systemic lupus erythematosus; ANCA associated vasculitis; Sjogren’s syndrome; B cell; T cell; Tyrosine kinase inhibitors; chemokine receptors; Cellular targeting INTRODUCTION There have been important advances in our understanding of the pathogenesis of autoimmune disease over the past two decades, which have led to an expanding array of biologic therapeutics targeting selective immune responses. -

Benlysta 200 Mg
This leaflet format has been determined by the Ministry of Health and the content has been checked and approved in December 2018 Benlysta 200 mg 1. NAME OF THE MEDICINAL PRODUCT Benlysta 200 mg solution for injection in pre-filled pen. Benlysta 200 mg solution for injection in pre-filled syringe. 2. QUALITATIVE AND QUANTITATIVE COMPOSITION Pre-filled pen Each 1-ml pre-filled pen contains 200 mg of belimumab. Pre-filled syringe Each 1-ml pre-filled syringe contains 200 mg of belimumab. Belimumab is a human, IgG1λ monoclonal antibody, produced in a mammalian cell line (NS0) by recombinant DNA technology. For the full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM Solution for injection in pre-filled pen (injection) Solution for injection in pre-filled syringe (injection) A clear to opalescent, colourless to pale yellow solution, with a pH of 6. 4. CLINICAL PARTICULARS 4.1 Therapeutic indications Benlysta is indicated as add-on therapy in adult patients with active, autoantibody-positive systemic lupus erythematosus (SLE) with a high degree of disease activity (e.g., positive anti-dsDNA and low complement) despite standard therapy (see section 5.1). 4.2 Posology and method of administration Benlysta treatment should be initiated and supervised by a qualified physician experienced in the diagnosis and treatment of SLE. It is recommended that the first subcutaneous injection of Benlysta should be under the supervision of a healthcare professional in a setting that is sufficiently qualified to manage hypersensitivity reactions, if necessary. The healthcare professional must provide proper training in subcutaneous technique and education about signs and symptoms of hypersensitivity reactions (see section 4.4). -

Belimumab (Benlysta)
Belimumab PATIENT FACT SHEET (Benlysta) Belimumab is the first drug designed to treat moderate lupus affecting the skin, joints, and other lupus. It was the first drug approved for lupus by the organs. It works against a protein that triggers certain FDA in more than 50 years. It is used in combination cells in the immune system to attack different parts with other lupus drugs, such as hydroxychloroquine of the body. and steroids. Belimumab treats people with mild or WHAT IS IT? Belimumab can be given as an infusion or injection. and is then given every four weeks. Usually, it takes effect When given by infusion, the medication is administered in 12 weeks, but it can take longer in some patients. through a needle that is inserted into one of the veins by Belimumab is also available in a self-injection form. This a health care professional. The infusion lasts about one means you can give it to yourself by injecting under the hour. The dose is adjusted to your weight. The infusion skin once a week. The dose for the self-injection is 200 mg HOW TO schedule starts at every two weeks for the first four weeks, TAKE IT and is pre-loaded into a syringe or device for you. Belimumab can cause certain side effects, although Belimumab can lower the number of white blood cells in many of these are mild. The most common side effects the blood of some people. It can also cause you to feel include headaches, diarrhea, nausea, muscle aches, down and have thoughts of harming yourself, especially and infections, such as colds, bronchitis, and urinary if you have a history of depression.