The Closest Unicellular Relatives of Animals
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

An Ichthyophonus Hoferi Epizootic in Herring in the North Sea, the Skagerrak, the Kattegat and the Baltic Sea
Downloaded from orbit.dtu.dk on: Oct 04, 2021 An Ichthyophonus hoferi epizootic in herring in the North Sea, the Skagerrak, the Kattegat and the Baltic Sea Mellergaard, Stig; Spanggaard, Bettina Published in: Diseases of Aquatic Organisms Link to article, DOI: 10.3354/dao028191 Publication date: 1997 Document Version Publisher's PDF, also known as Version of record Link back to DTU Orbit Citation (APA): Mellergaard, S., & Spanggaard, B. (1997). An Ichthyophonus hoferi epizootic in herring in the North Sea, the Skagerrak, the Kattegat and the Baltic Sea. Diseases of Aquatic Organisms, 28(3), 191-199. https://doi.org/10.3354/dao028191 General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. Users may download and print one copy of any publication from the public portal for the purpose of private study or research. You may not further distribute the material or use it for any profit-making activity or commercial gain You may freely distribute the URL identifying the publication in the public portal If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. DISEASES OF AQUATIC ORGANISMS Vol. 28: 191-199, 1997 Published March 27 Dis Aquat Org 1 An Ichthyophonus hoferi epizootic in herring in the North Sea, the Skagerrak, the Kattegat and the Baltic Sea 'Danish Institute of Fisheries Research, Department for Marine and Coastal Ecology. -
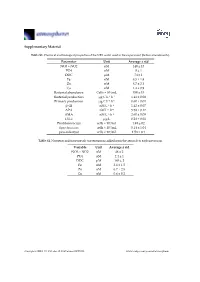
Supplementary Material Parameter Unit Average ± Std NO3 + NO2 Nm
Supplementary Material Table S1. Chemical and biological properties of the NRS water used in the experiment (before amendments). Parameter Unit Average ± std NO3 + NO2 nM 140 ± 13 PO4 nM 8 ± 1 DOC μM 74 ± 1 Fe nM 8.5 ± 1.8 Zn nM 8.7 ± 2.1 Cu nM 1.4 ± 0.9 Bacterial abundance Cells × 104/mL 350 ± 15 Bacterial production μg C L−1 h−1 1.41 ± 0.08 Primary production μg C L−1 h−1 0.60 ± 0.01 β-Gl nM L−1 h−1 1.42 ± 0.07 APA nM L−1 h−1 5.58 ± 0.17 AMA nM L−1·h−1 2.60 ± 0.09 Chl-a μg/L 0.28 ± 0.01 Prochlorococcus cells × 104/mL 1.49 ± 02 Synechococcus cells × 104/mL 5.14 ± 1.04 pico-eukaryot cells × 103/mL 1.58 × 0.1 Table S2. Nutrients and trace metals concentrations added from the aerosols to each mesocosm. Variable Unit Average ± std NO3 + NO2 nM 48 ± 2 PO4 nM 2.4 ± 1 DOC μM 165 ± 2 Fe nM 2.6 ± 1.5 Zn nM 6.7 ± 2.5 Cu nM 0.6 ± 0.2 Atmosphere 2019, 10, 358; doi:10.3390/atmos10070358 www.mdpi.com/journal/atmosphere Atmosphere 2019, 10, 358 2 of 6 Table S3. ANOVA test results between control, ‘UV-treated’ and ‘live-dust’ treatments at 20 h or 44 h, with significantly different values shown in bold. ANOVA df Sum Sq Mean Sq F Value p-value Chl-a 20 H 2, 6 0.03, 0.02 0.02, 0 4.52 0.0634 44 H 2, 6 0.02, 0 0.01, 0 23.13 0.002 Synechococcus Abundance 20 H 2, 7 8.23 × 107, 4.11 × 107 4.11 × 107, 4.51 × 107 0.91 0.4509 44 H 2, 7 5.31 × 108, 6.97 × 107 2.65 × 108, 1.16 × 107 22.84 0.0016 Prochlorococcus Abundance 20 H 2, 8 4.22 × 107, 2.11 × 107 2.11 × 107, 2.71 × 106 7.77 0.0216 44 H 2, 8 9.02 × 107, 1.47 × 107 4.51 × 107, 2.45 × 106 18.38 0.0028 Pico-eukaryote -

Predatory Flagellates – the New Recently Discovered Deep Branches of the Eukaryotic Tree and Their Evolutionary and Ecological Significance
Protistology 14 (1), 15–22 (2020) Protistology Predatory flagellates – the new recently discovered deep branches of the eukaryotic tree and their evolutionary and ecological significance Denis V. Tikhonenkov Papanin Institute for Biology of Inland Waters, Russian Academy of Sciences, Borok, 152742, Russia | Submitted March 20, 2020 | Accepted April 6, 2020 | Summary Predatory protists are poorly studied, although they are often representing important deep-branching evolutionary lineages and new eukaryotic supergroups. This short review/opinion paper is inspired by the recent discoveries of various predatory flagellates, which form sister groups of the giant eukaryotic clusters on phylogenetic trees, and illustrate an ancestral state of one or another supergroup of eukaryotes. Here we discuss their evolutionary and ecological relevance and show that the study of such protists may be essential in addressing previously puzzling evolutionary problems, such as the origin of multicellular animals, the plastid spread trajectory, origins of photosynthesis and parasitism, evolution of mitochondrial genomes. Key words: evolution of eukaryotes, heterotrophic flagellates, mitochondrial genome, origin of animals, photosynthesis, predatory protists, tree of life Predatory flagellates and diversity of eu- of the hidden diversity of protists (Moon-van der karyotes Staay et al., 2000; López-García et al., 2001; Edg- comb et al., 2002; Massana et al., 2004; Richards The well-studied multicellular animals, plants and Bass, 2005; Tarbe et al., 2011; de Vargas et al., and fungi immediately come to mind when we hear 2015). In particular, several prevailing and very abun- the term “eukaryotes”. However, these groups of dant ribogroups such as MALV, MAST, MAOP, organisms represent a minority in the real diversity MAFO (marine alveolates, stramenopiles, opistho- of evolutionary lineages of eukaryotes. -

S41467-021-25308-W.Pdf
ARTICLE https://doi.org/10.1038/s41467-021-25308-w OPEN Phylogenomics of a new fungal phylum reveals multiple waves of reductive evolution across Holomycota ✉ ✉ Luis Javier Galindo 1 , Purificación López-García 1, Guifré Torruella1, Sergey Karpov2,3 & David Moreira 1 Compared to multicellular fungi and unicellular yeasts, unicellular fungi with free-living fla- gellated stages (zoospores) remain poorly known and their phylogenetic position is often 1234567890():,; unresolved. Recently, rRNA gene phylogenetic analyses of two atypical parasitic fungi with amoeboid zoospores and long kinetosomes, the sanchytrids Amoeboradix gromovi and San- chytrium tribonematis, showed that they formed a monophyletic group without close affinity with known fungal clades. Here, we sequence single-cell genomes for both species to assess their phylogenetic position and evolution. Phylogenomic analyses using different protein datasets and a comprehensive taxon sampling result in an almost fully-resolved fungal tree, with Chytridiomycota as sister to all other fungi, and sanchytrids forming a well-supported, fast-evolving clade sister to Blastocladiomycota. Comparative genomic analyses across fungi and their allies (Holomycota) reveal an atypically reduced metabolic repertoire for sanchy- trids. We infer three main independent flagellum losses from the distribution of over 60 flagellum-specific proteins across Holomycota. Based on sanchytrids’ phylogenetic position and unique traits, we propose the designation of a novel phylum, Sanchytriomycota. In addition, our results indicate that most of the hyphal morphogenesis gene repertoire of multicellular fungi had already evolved in early holomycotan lineages. 1 Ecologie Systématique Evolution, CNRS, Université Paris-Saclay, AgroParisTech, Orsay, France. 2 Zoological Institute, Russian Academy of Sciences, St. ✉ Petersburg, Russia. 3 St. -

Group of Microorganisms at the Animal-Fungal Boundary
16 Aug 2002 13:56 AR AR168-MI56-14.tex AR168-MI56-14.SGM LaTeX2e(2002/01/18) P1: GJC 10.1146/annurev.micro.56.012302.160950 Annu. Rev. Microbiol. 2002. 56:315–44 doi: 10.1146/annurev.micro.56.012302.160950 First published online as a Review in Advance on May 7, 2002 THE CLASS MESOMYCETOZOEA: A Heterogeneous Group of Microorganisms at the Animal-Fungal Boundary Leonel Mendoza,1 John W. Taylor,2 and Libero Ajello3 1Medical Technology Program, Department of Microbiology and Molecular Genetics, Michigan State University, East Lansing Michigan, 48824-1030; e-mail: [email protected] 2Department of Plant and Microbial Biology, University of California, Berkeley, California 94720-3102; e-mail: [email protected] 3Centers for Disease Control and Prevention, Mycotic Diseases Branch, Atlanta Georgia 30333; e-mail: [email protected] Key Words Protista, Protozoa, Neomonada, DRIP, Ichthyosporea ■ Abstract When the enigmatic fish pathogen, the rosette agent, was first found to be closely related to the choanoflagellates, no one anticipated finding a new group of organisms. Subsequently, a new group of microorganisms at the boundary between an- imals and fungi was reported. Several microbes with similar phylogenetic backgrounds were soon added to the group. Interestingly, these microbes had been considered to be fungi or protists. This novel phylogenetic group has been referred to as the DRIP clade (an acronym of the original members: Dermocystidium, rosette agent, Ichthyophonus, and Psorospermium), as the class Ichthyosporea, and more recently as the class Mesomycetozoea. Two orders have been described in the mesomycetozoeans: the Der- mocystida and the Ichthyophonida. So far, all members in the order Dermocystida have been pathogens either of fish (Dermocystidium spp. -

Effects of Ichthyophonus on Survival and Reproductive Success of Yukon River Chinook Salmon
U.S. Fish and Wildlife Service Office of Subsistence Management Fisheries Resource Monitoring Program Effects of Ichthyophonus on Survival and Reproductive Success of Yukon River Chinook Salmon Final Report for Study 01-200 Richard Kocan and Paul Hershberger* School of Aquatic & Fishery Sciences, Box 355100 University of Washington, Seattle, WA 98195 Phone: 206-685-3275 e-mail: [email protected] and James Winton Western Fisheries Research Center, USGS-BRD, 6505 NE 65th Street, Seattle, WA 98115 Phone: 206-526-6587 e-mail: [email protected] July 2004 *Present Address: Marrowstone Marine Station, USGS-BRD, 616 Marrowstone Point Road, Nordland, WA 98358; Phone: 360-385-1007; e-mail: [email protected] TABLE OF CONTENTS Abstract, keywords, and citation.……………………..……………………………. 4 Introduction………………………………………………………………………… 5 Objectives………………………………………………………………………….. 6 Methods………………………………………………….…………………………. 6 Results……………………………………………………..……………………….. 13 Discussion………………………………………………………………………….. 17 Summary…………………………………………………………………………… 24 Conclusions………………………………………………………………………… 24 Acknowledgements………………………………………………………………… 25 Literature cited……………………………………………..………………………. 25 Footnotes…………………………………………………………………………… 29 Figures Figure 1 Map of Alaska showing sample sites along the Yukon and Tanana Rivers………………………….……………….………………….… 30 Figure 2 Infection prevalence in male and female chinook salmon from the Yukon River mainstem all years combined.……….………….……. 31 Figure 3 Annual infection prevalence 1999-2002.…………………………… 32 Figure 4 Ichthyophonus infection -

Microbial Eukaryotes in Oil Sands Environments: Heterotrophs in the Spotlight
microorganisms Review Microbial Eukaryotes in Oil Sands Environments: Heterotrophs in the Spotlight Elisabeth Richardson 1,* and Joel B. Dacks 2,* 1 Department of Biological Sciences, University of Alberta, Edmonton, AB T6G 2E9, Canada 2 Division of Infectious Disease, Department of Medicine, University of Alberta, Edmonton, AB T6G 2G3, Canada * Correspondence: [email protected] (E.R.); [email protected] (J.B.D.); Tel.: +1-780-248-1493 (J.B.D.) Received: 6 May 2019; Accepted: 14 June 2019; Published: 19 June 2019 Abstract: Hydrocarbon extraction and exploitation is a global, trillion-dollar industry. However, for decades it has also been known that fossil fuel usage is environmentally detrimental; the burning of hydrocarbons results in climate change, and environmental damage during extraction and transport can also occur. Substantial global efforts into mitigating this environmental disruption are underway. The global petroleum industry is moving more and more into exploiting unconventional oil reserves, such as oil sands and shale oil. The Albertan oil sands are one example of unconventional oil reserves; this mixture of sand and heavy bitumen lying under the boreal forest of Northern Alberta represent one of the world’s largest hydrocarbon reserves, but extraction also requires the disturbance of a delicate northern ecosystem. Considerable effort is being made by various stakeholders to mitigate environmental impact and reclaim anthropogenically disturbed environments associated with oil sand extraction. In this review, we discuss the eukaryotic microbial communities associated with the boreal ecosystem and how this is affected by hydrocarbon extraction, with a particular emphasis on the reclamation of tailings ponds, where oil sands extraction waste is stored. -

D070p001.Pdf
DISEASES OF AQUATIC ORGANISMS Vol. 70: 1–36, 2006 Published June 12 Dis Aquat Org OPENPEN ACCESSCCESS FEATURE ARTICLE: REVIEW Guide to the identification of fish protozoan and metazoan parasites in stained tissue sections D. W. Bruno1,*, B. Nowak2, D. G. Elliott3 1FRS Marine Laboratory, PO Box 101, 375 Victoria Road, Aberdeen AB11 9DB, UK 2School of Aquaculture, Tasmanian Aquaculture and Fisheries Institute, CRC Aquafin, University of Tasmania, Locked Bag 1370, Launceston, Tasmania 7250, Australia 3Western Fisheries Research Center, US Geological Survey/Biological Resources Discipline, 6505 N.E. 65th Street, Seattle, Washington 98115, USA ABSTRACT: The identification of protozoan and metazoan parasites is traditionally carried out using a series of classical keys based upon the morphology of the whole organism. However, in stained tis- sue sections prepared for light microscopy, taxonomic features will be missing, thus making parasite identification difficult. This work highlights the characteristic features of representative parasites in tissue sections to aid identification. The parasite examples discussed are derived from species af- fecting finfish, and predominantly include parasites associated with disease or those commonly observed as incidental findings in disease diagnostic cases. Emphasis is on protozoan and small metazoan parasites (such as Myxosporidia) because these are the organisms most likely to be missed or mis-diagnosed during gross examination. Figures are presented in colour to assist biologists and veterinarians who are required to assess host/parasite interactions by light microscopy. KEY WORDS: Identification · Light microscopy · Metazoa · Protozoa · Staining · Tissue sections Resale or republication not permitted without written consent of the publisher INTRODUCTION identifying the type of epithelial cells that compose the intestine. -

Common Diseases of Wild and Cultured Fishes in Alaska
COMMON DISEASES OF WILD AND CULTURED FISHES IN ALASKA Theodore Meyers, Tamara Burton, Collette Bentz and Norman Starkey July 2008 Alaska Department of Fish and Game Fish Pathology Laboratories The Alaska Department of Fish and Game printed this publication at a cost of $12.03 in Anchorage, Alaska, USA. 3 About This Booklet This booklet is a product of the Ichthyophonus Diagnostics, Educational and Outreach Program which was initiated and funded by the Yukon River Panel’s Restoration and Enhancement fund and facilitated by the Yukon River Drainage Fisheries Association in conjunction with the Alaska Department of Fish and Game. The original impetus driving the production of this booklet was from a concern that Yukon River fishers were discarding Canadian-origin Chinook salmon believed to be infected by Ichthyophonus. It was decided to develop an educational program that included the creation of a booklet containing photographs and descriptions of frequently encountered parasites within Yukon River fish. This booklet is to serve as a brief illustrated guide that lists many of the common parasitic, infectious, and noninfectious diseases of wild and cultured fish encountered in Alaska. The content is directed towards lay users, as well as fish culturists at aquaculture facilities and field biologists and is not a comprehensive treatise nor should it be considered a scientific document. Interested users of this guide are directed to the listed fish disease references for additional information. Information contained within this booklet is published from the laboratory records of the Alaska Department of Fish and Game, Fish Pathology Section that has regulatory oversight of finfish health in the State of Alaska. -

Tracing Evolutionary Ages of Cancer-Driving Sites by Cancer
bioRxiv preprint doi: https://doi.org/10.1101/2020.02.09.940528; this version posted February 10, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Tracing Evolutionary Ages of Cancer-Driving Sites by Cancer Somatic Mutations Xun Gu*1, Zhan Zou2, Jingwen Yang3,4 1Department of Genetics, Development and Cell Biology, Iowa State University, Ames, IA 50011, USA. 2College of Pharmaceutical Sciences, Zhejiang University, Hangzhou 301158, China 3School of Life Sciences, Fudan University, Shanghai 200433, China. 4Human Phenome Institute, Fudan University, Shanghai 200433, China. *Corresponding author. E-mail: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.02.09.940528; this version posted February 10, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract Evolutionary understanding of cancer genes may provide insights on the nature and evolution of complex life and the origin of multicellularity. In this study, we focus on the evolutionary ages of cancer-driving sites, and try to explore to what extent the amino acids of cancer-driving sites can be traced back to the most recent common ancestor (MRCA) of the gene. According to gene phylostraigraphy analysis, we use the definition of gene age (tg) by the most ancient phylogenetic position that can be traced back, in most cases based on the large-scale homology search of protein sequences. -

Molecular Phylogeny of Choanoflagellates, the Sister Group to Metazoa
Molecular phylogeny of choanoflagellates, the sister group to Metazoa M. Carr*†, B. S. C. Leadbeater*‡, R. Hassan‡§, M. Nelson†, and S. L. Baldauf†¶ʈ †Department of Biology, University of York, Heslington, York, YO10 5YW, United Kingdom; and ‡School of Biosciences, University of Birmingham, Edgbaston, Birmingham, B15 2TT, United Kingdom Edited by Andrew H. Knoll, Harvard University, Cambridge, MA, and approved August 28, 2008 (received for review February 28, 2008) Choanoflagellates are single-celled aquatic flagellates with a unique family. Members of the Acanthoecidae family (Norris 1965) are morphology consisting of a cell with a single flagellum surrounded by characterized by the most distinct periplast morphology. This a ‘‘collar’’ of microvilli. They have long interested evolutionary biol- consists of a complex basket-like lorica constructed in a precise and ogists because of their striking resemblance to the collared cells highly reproducible manner from ribs (costae) composed of rod- (choanocytes) of sponges. Molecular phylogeny has confirmed a close shaped silica strips (Fig. 1 E and F) (13). The Acanthoecidae family relationship between choanoflagellates and Metazoa, and the first is further subdivided into nudiform (Fig. 1E) and tectiform (Fig. 1F) choanoflagellate genome sequence has recently been published. species, based on the morphology of the lorica, the stage in the cell However, molecular phylogenetic studies within choanoflagellates cycle when the silica strips are produced, the location at which the are still extremely limited. Thus, little is known about choanoflagel- strips are stored, and the mode of cell division [supporting infor- late evolution or the exact nature of the relationship between mation (SI) Text] (14). -

An Unusual Form of Ichthyophonus Hoferi (Ichthyophonales: Ichthyophonaceae) from Yellowtail Flounder Limanda Ferruginea from the Nova Scotia Shelf
DISEASES OF AQUATIC ORGANISMS Vol. 18: 21-28.1994 Published January 27 Dis. aquat. Org. I An unusual form of Ichthyophonus hoferi (Ichthyophonales: Ichthyophonaceae) from yellowtail flounder Limanda ferruginea from the Nova Scotia shelf Thomas G. Rand Biology Department, Saint Mary's University, Halifax, Nova Scotia, Canada B3H 3C3 ABSTRACT: An unusual form of Ichthyophonus hoferi is described from 12 of 254 (4.7 %) yellowtail flounder Limanda ferruginea Storer sampled from Brown's Bank on the Nova Scotia shelf This patho- gen was the cause of focal, circular to lobate, creamy-white lesions on the liver and kidneys of infected fishes. By virtue of its morphological and dimensional characteristics in the fish tissues, the histochem- ical profile of its thallus walls, and its development in vitro, this form was easily distinguished from I. hoferi sensu Plehn & Mulsow, 1911 from yellowtail flounder from other locations on the Nova Scotia shelf, and from other freshwater and marine fish species. However, gross signs of the infection, as well as the morphological, dimensional, andor histochemical features of this form of I. hoferi were so re- markably similar to those of an 'I. hoferi pathogen from L. ferruginea, Myxocephalus octodecemspino- sus, and Scomber scombrus described in the literature, and to an Ichthyophonus-type pathogen from Scopelogadus beanii, as to suggest that they are the same. KEY WORDS: Ichthyophonus hoferi . Yellowtail flounder. Northwest Atlantic . Limanda ferrugjnea INTRODUCTION effects and that the reported forms are a single species, namely Ichthyophonus hoferi sensu Plehn & Mulsow, Ichthyophonus hoferi Plehn & Mulsow, 1911 has 1911. However, Alderman (1976, 1982), Johnson & been reported from a wide variety of host species Sparrow (1961), MacKenzie (1979), and Neish & including crustaceans (see Reichenbach-Klinke 1957, Hughes (1980) have suggested that the genus Sindermann 1970), fishes (see Reichenbach-Klincke Ichthyophonus and especially I.