Identi Cation of a Methylation-Driven Gene Panel for Survival Prediction
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Novel Candidate Key Drivers in the Integrative Network of Genes, Micrornas, Methylations, and Copy Number Variations in Squamous Cell Lung Carcinoma
Hindawi Publishing Corporation BioMed Research International Volume 2015, Article ID 358125, 11 pages http://dx.doi.org/10.1155/2015/358125 Research Article Novel Candidate Key Drivers in the Integrative Network of Genes, MicroRNAs, Methylations, and Copy Number Variations in Squamous Cell Lung Carcinoma Tao Huang,1,2 Jing Yang,2 and Yu-dong Cai1 1 CollegeofLifeScience,ShanghaiUniversity,Shanghai200444,China 2Institute of Health Sciences, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences and Shanghai Jiao Tong University School of Medicine, Shanghai 200031, China Correspondence should be addressed to Tao Huang; [email protected] and Yu-dong Cai; cai [email protected] Received 17 September 2014; Revised 6 January 2015; Accepted 22 January 2015 Academic Editor: Aparup Das Copyright © 2015 Tao Huang et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. The mechanisms of lung cancer are highly complex. Not only mRNA gene expression but also microRNAs, DNA methylation, and copy number variation (CNV) play roles in tumorigenesis. It is difficult to incorporate so much information into a single model that can comprehensively reflect all these lung cancer mechanisms. In this study, we analyzed the 129 TCGA (The Cancer Genome Atlas) squamous cell lung carcinoma samples with gene expression, microRNA expression, DNA methylation, and CNV data. First, we used variance inflation factor (VIF) regression to build the whole genome integrative network. Then, we isolated the lung cancer subnetwork by identifying the known lung cancer genes and their direct regulators. -

Autophagy Modulation in Bladder Cancer Development and Treatment (Review)
ONCOLOGY REPORTS 42: 1647-1655, 2019 Autophagy modulation in bladder cancer development and treatment (Review) FAPING LI, HUI GUO, YUXUAN YANG, MINGLIANG FENG, BIN LIU, XIANG REN and HONGLAN ZHOU Department of Urology, The First Hospital of Jilin University, Changchun, Jilin 130021, P.R. China Received April 24, 2019; Accepted August 1, 2019 DOI: 10.3892/or.2019.7286 Abstract. Bladder cancer (BC) is a potentially life-threatening 1. Introduction malignancy. Due to a high recurrence rate, frequent surveil- lance strategies and intravesical drug therapies, BC is Bladder cancer (BC) is a potentially life-threatening malig- considered one of the most expensive tumors to treat. As a nancy that is considered one of the most expensive tumors fundamental evolutionary catabolic process, autophagy plays in terms of treatment and medical care (1-3). After prostate an important role in the maintenance of cellular environ- cancer, it is the second most common type of urological cancer mental homeostasis by degrading and recycling damaged and ranks 10th among the most common types of cancer cytoplasmic components, including macromolecules and around the globe (4). It has been estimated that there were organelles. Scientific studies in the last two decades have 549,393 new cases of BC and 199,922 deaths resulting from shown that autophagy acts as a double‑edged sword with this disease worldwide in 2018, according to a report from regard to the treatment of cancer. On one hand, autophagy the International Agency for Research on Cancer (4). The inhibition is able to increase the sensitivity of cancer cells to primary histological subtype of human BC is transitional cell treatment, a process known as protective autophagy. -

Identification of C3 As a Therapeutic Target for Diabetic Nephropathy By
www.nature.com/scientificreports OPEN Identifcation of C3 as a therapeutic target for diabetic nephropathy by bioinformatics analysis ShuMei Tang, XiuFen Wang, TianCi Deng, HuiPeng Ge & XiangCheng Xiao* The pathogenesis of diabetic nephropathy is not completely understood, and the efects of existing treatments are not satisfactory. Various public platforms already contain extensive data for deeper bioinformatics analysis. From the GSE30529 dataset based on diabetic nephropathy tubular samples, we identifed 345 genes through diferential expression analysis and weighted gene coexpression correlation network analysis. GO annotations mainly included neutrophil activation, regulation of immune efector process, positive regulation of cytokine production and neutrophil-mediated immunity. KEGG pathways mostly included phagosome, complement and coagulation cascades, cell adhesion molecules and the AGE-RAGE signalling pathway in diabetic complications. Additional datasets were analysed to understand the mechanisms of diferential gene expression from an epigenetic perspective. Diferentially expressed miRNAs were obtained to construct a miRNA-mRNA network from the miRNA profles in the GSE57674 dataset. The miR-1237-3p/SH2B3, miR-1238-5p/ ZNF652 and miR-766-3p/TGFBI axes may be involved in diabetic nephropathy. The methylation levels of the 345 genes were also tested based on the gene methylation profles of the GSE121820 dataset. The top 20 hub genes in the PPI network were discerned using the CytoHubba tool. Correlation analysis with GFR showed that SYK, CXCL1, LYN, VWF, ANXA1, C3, HLA-E, RHOA, SERPING1, EGF and KNG1 may be involved in diabetic nephropathy. Eight small molecule compounds were identifed as potential therapeutic drugs using Connectivity Map. It is estimated that a total of 451 million people sufered from diabetes by 2017, and the number is speculated to be 693 million by 2045 1. -

GVES: Machine Learning Model for Identification of Prognostic Genes
www.nature.com/scientificreports OPEN GVES: machine learning model for identifcation of prognostic genes with a small dataset Soohyun Ko1, Jonghwan Choi2 & Jaegyoon Ahn1* Machine learning may be a powerful approach to more accurate identifcation of genes that may serve as prognosticators of cancer outcomes using various types of omics data. However, to date, machine learning approaches have shown limited prediction accuracy for cancer outcomes, primarily owing to small sample numbers and relatively large number of features. In this paper, we provide a description of GVES (Gene Vector for Each Sample), a proposed machine learning model that can be efciently leveraged even with a small sample size, to increase the accuracy of identifcation of genes with prognostic value. GVES, an adaptation of the continuous bag of words (CBOW) model, generates vector representations of all genes for all samples by leveraging gene expression and biological network data. GVES clusters samples using their gene vectors, and identifes genes that divide samples into good and poor outcome groups for the prediction of cancer outcomes. Because GVES generates gene vectors for each sample, the sample size efect is reduced. We applied GVES to six cancer types and demonstrated that GVES outperformed existing machine learning methods, particularly for cancer datasets with a small number of samples. Moreover, the genes identifed as prognosticators were shown to reside within a number of signifcant prognostic genetic pathways associated with pancreatic cancer. Te accurate identifcation of genes with prognostic value in the prediction of cancer outcomes is a challenging task for cancer researchers. Numerous statistical and computational methods have been developed to increase the accuracy of cancer prognosis1. -

Overexpression of Autocrine Motility Factor in Metastatic Tumor Cells: Possible Association with Augmented Expression of KIF3A and GDI-B
Laboratory Investigation (2004) 84, 513–522 & 2004 USCAP, Inc All rights reserved 0023-6837/04 $25.00 www.laboratoryinvestigation.org Overexpression of autocrine motility factor in metastatic tumor cells: possible association with augmented expression of KIF3A and GDI-b Takashi Yanagawa1, Hideomi Watanabe1, Toshiyuki Takeuchi2, Shuhei Fujimoto3, Hideyuki Kurihara2,4 and Kenji Takagishi1 1Department of Orthopaedic Surgery; 2Department of Molecular Medicine; 3Department of Microbiology and 4Department of Neurosurgery Gunma University Faculty of Medicine, Institute for Molecular and Cellular Regulation, Gunma University, Showa, Maebashi, Gunma 371-8511, Japan Autocrine motility factor (AMF), which is identical to phosphohexose isomerase (PHI)/glucose-6-phosphate isomerase (GPI) , a ubiquitous enzyme essential for glycolysis, neuroleukin (NLK), a neurotrophic growth factor, and maturation factor (MF) mediating the differentiation of human myeloid cells, enhances the motility and metastatic ability of tumor cells. AMF/PHI activity is elevated in the serum or urine in patients with malignant tumors. Here, we constructed an amf/phi/nlk/mf gene using adenovirus vector and transfected into two tumor cell lines. Overexpression of AMF/PHI/NLK/MF enhanced AMF secretion into the culture media in both tumor cell lines. However, upregulation of motility and metastatic ability was found only in metastatic fibrosarcoma cells expressing an AMF receptor, gp78, and was not found in gp78-undetectable osteosarcoma cells. Thus, not only serum AMF activity but also gp78-expression in tumor cells may be required for metastasis-related motility induction. With the use of microarray analyses, we detected two augmented genes, rho GDP dissociation inhibitor beta and kinesin motor 3A, as well as AMF itself. -

Mir-34/449 Control Apical Actin Network Formation During Multiciliogenesis Through Small Gtpase Pathways
ARTICLE Received 7 Jul 2015 | Accepted 17 Aug 2015 | Published 18 Sep 2015 DOI: 10.1038/ncomms9386 OPEN miR-34/449 control apical actin network formation during multiciliogenesis through small GTPase pathways Benoıˆt Chevalier1,2,*, Anna Adamiok3,*, Olivier Mercey1,2,*, Diego R. Revinski3, Laure-Emmanuelle Zaragosi1,2, Andrea Pasini3, Laurent Kodjabachian3, Pascal Barbry1,2 & Brice Marcet1,2 Vertebrate multiciliated cells (MCCs) contribute to fluid propulsion in several biological processes. We previously showed that microRNAs of the miR-34/449 family trigger MCC differentiation by repressing cell cycle genes and the Notch pathway. Here, using human and Xenopus MCCs, we show that beyond this initial step, miR-34/449 later promote the assembly of an apical actin network, required for proper basal bodies anchoring. Identification of miR-34/449 targets related to small GTPase pathways led us to characterize R-Ras as a key regulator of this process. Protection of RRAS messenger RNA against miR-34/449 binding impairs actin cap formation and multiciliogenesis, despite a still active RhoA. We propose that miR-34/449 also promote relocalization of the actin binding protein Filamin-A, a known RRAS interactor, near basal bodies in MCCs. Our study illustrates the intricate role played by miR-34/449 in coordinating several steps of a complex differentiation programme by regulating distinct signalling pathways. 1 CNRS, Institut de Pharmacologie Mole´culaire et Cellulaire (IPMC), UMR-7275, 660 route des Lucioles, 06560 Sophia-Antipolis, France. 2 University of Nice-Sophia-Antipolis (UNS), Institut de Pharmacologie Mole´culaire et Cellulaire, 660 route des Lucioles, Valbonne, 06560 Sophia-Antipolis, France. -
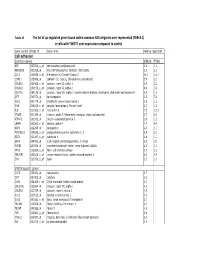
Tables 1S and 2S Mod 4June10
Table 1S The list of up‐regulated genes found within common GO catrgories over‐represented (FDR<0.1) in cells with TWIST1 over‐expression compared to control Gene symbol Probe ID Gene name Fold up-regulation Cell adhesion Common genes SNB19 T98G APC 203526_s_at adenomatosis polyposis coli 1.6 1.6 ARHGDIB 201288_at Rho GDP dissociation inhibitor (GDI) beta 2.1 2.1 CCL2 216598_s_at chemokine (C-C motif) ligand 2 44.0 16.7 CDH11 215608_at cadherin 11, type 2, OB-cadherin (osteoblast) 5.9 2.3 COL6A1 213428_s_at collagen, type VI, alpha 1 2.9 2.2 COL6A2 209156_s_at collagen, type VI, alpha 2 8.9 3.4 COL7A1 204136_at collagen, type VII, alpha 1 (epidermolysis bullosa, dystrophic, dominant and recessive) 2.8 1.6 DPT 213071_at dermatopontin 4.2 5.4 ECE1 201749_at endothelin converting enzyme 1 1.6 1.6 GSN 214040_s_at gelsolin (amyloidosis, Finnish type) 2.7 1.8 IL8 211506_s_at interleukin 8 7.5 12.1 ITGA5 201389_at integrin, alpha 5 (fibronectin receptor, alpha polypeptide) 1.7 2.6 KIFAP3 203333_at kinesin-associated protein 3 1.6 1.9 LAMA4 202202_s_at laminin, alpha 4 7.7 4.8 NRP1 212298_at neuropilin 1 2.6 1.7 PCDHGC3 215836_s_at protocadherin gamma subfamily C, 3 3.5 2.3 SDC1 201287_s_at syndecan 1 1.8 1.7 SRPX 204955_at sushi-repeat-containing protein, X-linked 5.0 3.5 TGFBI 201506_at transforming growth factor, beta-induced, 68kDa 2.5 1.6 THY1 213869_x_at Thy-1 cell surface antigen 1.7 2.2 TNFAIP6 206026_s_at tumor necrosis factor, alpha-induced protein 6 6.3 5.9 ZYX 215706_x_at zyxin 1.5 1.6 SNB19 specific genes CAPS 226424_at calcyphosine -

The Human Gene Connectome As a Map of Short Cuts for Morbid Allele Discovery
The human gene connectome as a map of short cuts for morbid allele discovery Yuval Itana,1, Shen-Ying Zhanga,b, Guillaume Vogta,b, Avinash Abhyankara, Melina Hermana, Patrick Nitschkec, Dror Friedd, Lluis Quintana-Murcie, Laurent Abela,b, and Jean-Laurent Casanovaa,b,f aSt. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY 10065; bLaboratory of Human Genetics of Infectious Diseases, Necker Branch, Paris Descartes University, Institut National de la Santé et de la Recherche Médicale U980, Necker Medical School, 75015 Paris, France; cPlateforme Bioinformatique, Université Paris Descartes, 75116 Paris, France; dDepartment of Computer Science, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel; eUnit of Human Evolutionary Genetics, Centre National de la Recherche Scientifique, Unité de Recherche Associée 3012, Institut Pasteur, F-75015 Paris, France; and fPediatric Immunology-Hematology Unit, Necker Hospital for Sick Children, 75015 Paris, France Edited* by Bruce Beutler, University of Texas Southwestern Medical Center, Dallas, TX, and approved February 15, 2013 (received for review October 19, 2012) High-throughput genomic data reveal thousands of gene variants to detect a single mutated gene, with the other polymorphic genes per patient, and it is often difficult to determine which of these being of less interest. This goes some way to explaining why, variants underlies disease in a given individual. However, at the despite the abundance of NGS data, the discovery of disease- population level, there may be some degree of phenotypic homo- causing alleles from such data remains somewhat limited. geneity, with alterations of specific physiological pathways under- We developed the human gene connectome (HGC) to over- come this problem. -

SUPPLEMENTARY MATERIAL Supplementary Fig. S1. LD Mice Used in This Study Accumulate Polyglucosan Inclusions (Lafora Bodies) in the Brain
1 SUPPLEMENTARY MATERIAL Supplementary Fig. S1. LD mice used in this study accumulate polyglucosan inclusions (Lafora bodies) in the brain. Samples from the hippocampus of five months old control, Epm2a-/- (lacking laforin) and Epm2b-/- mice (lacking malin) were stained with periodic acid Schiff reagent (PAS staining), which colors polysaccharide granules in red. Bar: 50 m. Supplementary Fig. S2. Principal component analysis (PCA) representing the first two components with the biggest level of phenotypic variability. Samples 1_S1 to 4_S4 corresponded to control, 5_S5, 6_S6 and 8_S8 to Epm2a-/- and 9_S9 to 12_S12 to Epm2b- /- samples, of animals of 16 months of age respectively. Supplementary Table S1. Primers used in this work to validate the expression of the corresponding genes by RT-qPCR. Supplementary Table S2: Genes downregulated more than 0.5 fold in Epm2a-/- and Epm2b-/- mice of 16 months of age. The gene name, false discovery rate (FDR), fold change (FC), description and MGI Id (mouse genome informatics) are indicated. Genes are arranged according to FC. Supplementary Table S3: Genes upregulated more than 1.5 fold in Epm2a-/- mice of 16 months of age. The gene name, false discovery rate (FDR), fold change (FC), description and MGI Id (mouse genome informatics) are indicated. Genes are arranged according to FC. Supplementary Table S4: Genes upregulated more than 1.5 fold in Epm2b-/- mice of 16 months of age. The gene name, false discovery rate (FDR), fold change (FC), description and MGI Id (mouse genome informatics) are indicated. Genes are arranged according to FC. 2 Supplementary Table S5: Genes upregulated in both Epm2a-/- and Epm2b-/- mice of 16 months of age. -

Regulation of Clustered Gene Expression by Cofactor of BRCA1 (COBRA1) in Breast Cancer Cells
Oncogene (2007) 26, 2543–2553 & 2007 Nature Publishing Group All rights reserved 0950-9232/07 $30.00 www.nature.com/onc ORIGINAL ARTICLE Regulation of clustered gene expression by cofactor of BRCA1 (COBRA1) in breast cancer cells SE Aiyar1, AL Blair1, DA Hopkinson, S Bekiranov and R Li Department of Biochemistry and Molecular Genetics, School of Medicine, University of Virginia, Charlottesville, VA, USA Eucaryotic genes that are coordinately expressed tend to the gene organization patterns within a genome. In be clustered. Furthermore, gene clusters across chromo- contrast to the long-held presumption that genes are somal regions are often upregulated in various tumors. randomly distributed throughout the eucaryotic ge- However, relatively little is known about how gene nomes, there is compelling evidence for clustering of clusters are coordinately expressed in physiological or co-expressed genes in every eucaryotic genome exam- pathological conditions. Cofactor of BRCA1 (COBRA1), ined so far (Hurst et al., 2004). For example, 25% of a subunit of the human negative elongation factor, has genes in budding yeast that are transcribed in the same been shown to repress estrogen-stimulated transcription cell-cycle stage are adjacently located (Cho et al., 1998). of trefoil factor 1 (TFF1 or pS2)by stalling RNA The gene-clustering phenomenon is perhaps even more polymerase II. Here, we carried out a genome-wide study prominent in multicellular organisms. It is reported that to identify additional physiological target genes of 20% of genes in the entire fly genome are organized into COBRA1 in breast cancer cells. The study identified a clusters with similar expression patterns (Spellman and total of 134 genes that were either activated or repressed Rubin, 2002). -
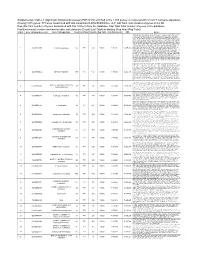
(FDR<0.05) Enriched in the 1015 Genes Co-Clustered with Known T
Supplementary Table 3. Significant biological processes (FDR<0.05) enriched in the 1,015 genes co-clustered with known T cell gene signatures. Among 1,015 genes, 771 were associated with GO annotation in DAVID database v6.7. List Total: total number of genes in my list. Pop Hits: total number of genes associated with this GO term from the database. Pop Total: total number of genes in the database. Fold Enrichment: relative enrichment ratio, calculated by (Count)/(List Total) divided by (Pop Hits)/(Pop Total). Index Gene Ontology Accession Gene Ontology Name Count List Total Pop Hits Pop Total Fold Enrichment FDR Genes AQP9, C1QC, B2M, LILRA1, LILRA2, CLEC4E, S1PR4, LILRA4, IFNG, LILRA6, CLEC4A, VNN1, ERAP2, FAS, CRTAM, C5AR1, GBP5, NCF2, NCF1, NCF4, SERPING1, HLA-DQA2, HLA-DQA1, PDCD1LG2, LILRB1, CCR9, C1QA, C1QB, LILRB2, CCR7, CCR6, UNC13D, CCR5, CD40LG, CCR4, LILRB3, CCR2, LILRB4, HLA-DPA1, VSIG4, HLA-DRA, IL1R2, IL1R1, HLA-DRB1, OAS3, ACP5, OAS1, OAS2, CD74, IFI35, ZAP70, FCER1G, HLA-DRB5, HLA-DPB1, HLA-DOA, HLA-DOB, DHX58, BLNK, IL23R, KIR2DS4, CD300C, SLAMF7, OASL, RGS1, APOL1, CD300A, HMHB1, CD209, CLEC7A, LY86, LY9, CLNK, FCRL4, SH2D1A, NOD2, HAMP, CCL3L1, CCL3L3, TICAM2, ICAM1, GZMA, CMKLR1, LY96, WAS, IL18BP, LAX1, TNFSF12- TNFSF13, HLA-DQB1, CSF2, GPR183, CCR1, GPR65, CXCL9, NCF1C, IL7R, CLEC10A, CCL24, CCL22, CYP27B1, CCL23, FCGR1C, FTHL3, FCGR1A, FCGR1B, BCL3, C2, CD27, CD28, FYB, IL18R1, IL7, CD1C, CTLA4, CCL19, CD1B, CD1A, TRIM22, CD180, CD1E, CCL18, CCL17, CCL13, FCGR2B, FCGR2C, P2RY14, LIME1, CD14, IL16, IL18, TLR1, TNFSF15, -

Sirnas in 786-O and Caki-1 Cells Was Detected by Reverse Transcrip- Tion-Quantitative Polymerase Chain Reaction
Figure S1. The silencing efficiency of (A) shRNAs and (B) siRNAs in 786-O and Caki-1 cells was detected by reverse transcrip- tion-quantitative polymerase chain reaction. (A) Relative PLEKHO1 expression in shRNA-transfected 786-O and Caki-1 cells. **P<0.01 vs. sh-Ctrl. (B) Relative PLEKHO1 expression in siRNA-transfected 786-O and Caki-1 cells. **P<0.001 vs. si-Ctrl. One-way analysis of variance followed by Dunnett’s multiple comparisons tests to compare differences among multiple groups. PLEKHO1, pleckstrin homology domain containing O1; sh, short hairpin; si, small interfering. Table SI. The file names to access for HTSeq - Counts data of the 527 RCC patients. id File name md5 Size State 001ae925-102c-4818-8eb0-c8d2e5726e7c d3f73c0f-d518-4e91-b038-a4360495ee27.htseq.counts.gz a20fd7085b4d1b35e228d65ae2cee800 255968 live 0085c623-bcdd-4b71-960c-a42acd0e53e4 801c2c60-5dc2-43f7-949f-18b5a35a4ca3.htseq.counts.gz 4bfe0e3c85512a673c822fa01763cb21 255034 live 00f072a6-6db0-4dcd-b819-e664f7e827df 9fa215ae-e9ce-4f87-9c8e-b3d9574925c8.htseq.counts.gz ec2d4e7a4d744f6698708daa4366c0a0 250343 live 02b2aa68-61fe-45af-8c2b-0e767df36cda 2fcd467f-4ea2-4712-ad16-217e05b135b1.htseq.counts.gz 959def0ef3c637316b50cb7928226bd0 251930 live 03d18e3c-0a38-482a-8ae0-5fa6906cb6f5 0048d446-301b-4d04-97d2-1348b6889692.htseq.counts.gz 158bbac7dc2235e4b0e784c72c5e8b27 258646 live 03fa41cb-b8b9-47ab-88d6-25852567da7b 476de4e4-7dfd-41a8-a3c5-3ae05987093e.htseq.counts.gz 4ee85f358d8f53b40e3cb48d922e9a99 249920 live 040b0c23-c706-41f1-8fa8-195509486588 2e5095b6-5f89-4b79-abfa-0afe326801ef.htseq.counts.gz