Environmental Protection Agency Pt. 63, App. A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Laboratory Glassware N Edition No
Laboratory Glassware n Edition No. 2 n Index Introduction 3 Ground joint glassware 13 Volumetric glassware 53 General laboratory glassware 65 Alphabetical index 76 Índice alfabético 77 Index Reference index 78 [email protected] Scharlau has been in the scientific glassware business for over 15 years Until now Scharlab S.L. had limited its sales to the Spanish market. However, now, coinciding with the inauguration of the new workshop next to our warehouse in Sentmenat, we are ready to export our scientific glassware to other countries. Standard and made to order Products for which there is regular demand are produced in larger Scharlau glassware quantities and then stocked for almost immediate supply. Other products are either manufactured directly from glass tubing or are constructed from a number of semi-finished products. Quality Even today, scientific glassblowing remains a highly skilled hand craft and the quality of glassware depends on the skill of each blower. Careful selection of the raw glass ensures that our final products are free from imperfections such as air lines, scratches and stones. You will be able to judge for yourself the workmanship of our glassware products. Safety All our glassware is annealed and made stress free to avoid breakage. Fax: +34 93 715 67 25 Scharlab The Lab Sourcing Group 3 www.scharlab.com Glassware Scharlau glassware is made from borosilicate glass that meets the specifications of the following standards: BS ISO 3585, DIN 12217 Type 3.3 Borosilicate glass ASTM E-438 Type 1 Class A Borosilicate glass US Pharmacopoeia Type 1 Borosilicate glass European Pharmacopoeia Type 1 Glass The typical chemical composition of our borosilicate glass is as follows: O Si 2 81% B2O3 13% Na2O 4% Al2O3 2% Glass is an inorganic substance that on cooling becomes rigid without crystallising and therefore it has no melting point as such. -
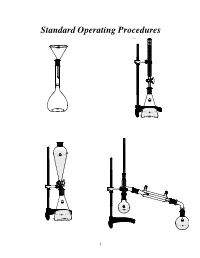
Standard Operating Procedures
Standard Operating Procedures 1 Standard Operating Procedures OVERVIEW In the following laboratory exercises you will be introduced to some of the glassware and tech- niques used by chemists to isolate components from natural or synthetic mixtures and to purify the individual compounds and characterize them by determining some of their physical proper- ties. While working collaboratively with your group members you will become acquainted with: a) Volumetric glassware b) Liquid-liquid extraction apparatus c) Distillation apparatus OBJECTIVES After finishing these sessions and reporting your results to your mentor, you should be able to: • Prepare solutions of exact concentrations • Separate liquid-liquid mixtures • Purify compounds by recrystallization • Separate mixtures by simple and fractional distillation 2 EXPERIMENT 1 Glassware Calibration, Primary and Secondary Standards, and Manual Titrations PART 1. Volumetric Glassware Calibration Volumetric glassware is used to either contain or deliver liquids at a specified temperature. Glassware manufacturers indicate this by inscribing on the volumetric ware the initials TC (to contain) or TD (to deliver) along with the calibration temperature, which is usually 20°C1. Volumetric glassware must be scrupulously clean before use. The presence of streaks or droplets is an indication of the presence of a grease film. To eliminate grease from glassware, scrub with detergent solution, rinse with tap water, and finally rinse with a small portion of distilled water. Volumetric flasks (TC) A volumetric flask has a large round bottom with only one graduation mark positioned on the long narrow neck. Graduation Mark Stopper The position of the mark facilitates the accurate and precise reading of the meniscus. If the flask is used to prepare a solution starting with a solid compound, add small amounts of sol- vent until the entire solid dissolves. -

Q & a – Accuracy and Precision
Food Analysis – FScN 4312W Laboratory: Assessment of Accuracy and Precision Key to Questions 1. Theoretically, how are standard deviation, coefficient of variation, mean, percent relative error, and 95% confidence interval affected by: (1) more replicates, and (2) a larger size of measurement? Was this evident in looking at the actual results obtained using the volumetric pipettes and the buret, with n = 3 vs n = 6, and with 1mL vs 10mL? Ans: (1) As the sample size increases (more replicates) - Calculated mean approaches the true mean - Standard deviation is inversely proportional to the square root of sample size, hence it decreases - CV decreases, as standard deviation approaches 0 for larger sample size - % Relative error decreases as calculated mean approaches true mean - 95% confidence interval narrows down (2) Larger size of measurement - Mean is close to true mean - Standard deviation might be larger due to larger size - CV decreases - % Relative error decreases, as calculated mean is close to true mean - 95% confidence interval narrows down Not all of the above assumptions were confirmed in this experiment according to the data observed. One reason could be that the sample size was very small (n = 3 and n = 6), to notice significant differences, another reason could be human error and variation from one person’s measuring technique to another. 2. Why are percent relative error and coefficient of variation used to compare the accuracy and precision, respectively, of the volumes from pipetting/dispensing 1 and 10mL with the volumetric pipettes and buret in parts 2 and 3, rather than simply the mean and standard deviation, respectively? Ans: Mean calculated from the readings gives the calculated mean, which may differ from the true mean. -

TOP-014, Technical Operating Procedure for Spectrophotometric
CENTER FOR NUCLEAR WASTE Proc. T0P-014 REGULATORY ANALYSES Revision ° TECHNICAL OPERATING PROCEDURE Page I of 11 Title TECHNICAL OPERATING PROCEDURE FOR SPECTROPHOTOMETRIC DETERMINATION OF SILICA EFFECTIVITY AND APPROVAL Revision n of this procedure became effective on 03/04/91 This procedure consists of the pages and changes listed below. Page No. Change Date Effective ALL 03/04/91 Superseaes Procedure No. None Approvals Written By Technical Revie / CENTER FOR NUCLEAR WASTE Proc.TOP-014 REGULATORY ANALYSES Revision _ TECHNICAL OPERATING PROCEDURE Page 2 of 1il TOP-014 TECHNICAL OPERATING PROCEDURE FOR SPECTROPHOTOMETRIC DETERMINATION OF SILICA 1. PURPOSE The purpose of this procedure is to describe a general method for the spectrophotometric determination of silica (Si02) in aqueous solutions. This procedure will be used for aqueous solutions generated by geochemical experiments involving silicate minerals (e.g., zeolites and feldspars) but may also be used for analyses of water samples collected from laboratory experiments or in the field. This procedure implements the requirements of CQAM Section 3. 2. APPLICABLE DOCUMENT The following document forms a part of this procedure, as applicable: Operator's Manual for Milton Roy Spectronic 1201 Spectrophotometer (1988) 3. RESPONSIBILITY (1) The Geosciences Element Manager shall be responsible for the development and maintainance of this procedure. (2) The cognizant Principal Investigator shall be responsible for the implementation of this procedure. (3) Personnel performing tasks described in this procedure are responsible for complying with its requirements. 4. EQUIPMENT (1) Milton Roy Spectronic 1201 Spectrophotometer with microprocessor-controlled functions and features including a linear curve fit test mode or equivalent. -

List of Equipment
Mfg & Exporters: All types of Turnkey Projects, , Beverages, Mineral Water plant & OEM Spares List Of Equipment SR.NO Equipment Qty 1. Autoclave 1 Vertical Complete Heavy gauze with pressure gauze, Safety Valve, Stream Release Valve Size 30 X 30 cm 2. Digital Hot Air Ageing oven 1 Size 12” x 12” x 12” made of MS powder coated. Make- HB 3. Digital pH System : 1 Make- HB 4. Digital Incubator Bacteriological 2 12” X 12” X 12” made of MS powder coated. Make- HB 5. Water bath double walled 1 inner chamber of SS outer M.S powder coated with 6 hole with thermostatic control Make- HB 6. INNOCULATION CHAMBER, M.S POWDER COATED WITH 1 UV & 1 FLOROCENT LIGHT INBUILT 1 Make- HB 7. Automatic Hot Plate with 8 inch dia with Thermostat 1 Make-HB 8. Digital Spectrophotometer 1 Make- HB 9. Digital Turbidity meter 1 Make- HB 10. Digital Colony Counter 1 Make- HB 11. S.S. Filter holder 47 mm 1 (complete S.S) 12. Microscope student 1 Make-Labron 13. Magnetic Stirrer with Hot Plate with 1 lit capacity Top made of SS with Teflon Bed 1 Make- HB 14. Centrifuge ( 4 holders) 1 Make- HB 15. Single pan Balance ( pocket type) max: 0-500 gm, L.C: 0.1 gm 1 16. High pressure vacuum Pump 1 17. BOD Incubators inner complete S.S outer MS powder coated.( heavy) 2 Make – HB 18. Heating Mental 1 lit capacity 1 Make- HB 19. S.S distillation Unit 1 Address: 140, Tribhuvan Estate opp Road No 11 Kathwada GIDC Contact: +91 9726943972 Email: [email protected] Mfg & Exporters: All types of Turnkey Projects, , Beverages, Mineral Water plant & OEM Spares List Of Chemicals Sr. -

ACP 2018:19 Platzinventarliste E Neu M. Bilder Grauton O Serienbrief
Institut für Anorganische Chemie - Platzinventarliste Raum T225 + T417 Center of Nanobionics and Photochemical Sciences (CNPS) Desk inventory room T225 + T417 Vorstand O.Univ.-Prof. DI Dr. Wolfgang Buchberger Desk Name, Matrikelnumber Adress Phone / Email Tel Name mob Matr# mail On the working bench 1 Water jet pump & non-return valve, hose øa*øi *l= 16*6*50 mm 1 Standbase w. frame rod 1 Tripod w. frame rod 1 Lab stool high, signed with number Upper lay 1 This Inventory (notlaminated yet) 1 Funnel Ø 40 mm 3 Filter crucible, glass, 1D3 1 Holder for separation funnel (2 sheets) 1 Glas rod l*Ø = 200*5mm 1 Thermometer 0-250°C + screw cap + ferrule 1 Volumetric pipette 10 ml with Rubber police man 2 Watch glas dish Ø 50 mm 1 Test tube, lateral tubing 1 Volumetric pipette 25 ml 2 Glas rod l*Ø = 200*8mm 2 Watch glas dish Ø 80 mm connector, 1 Graduated pipette 5 ml 1 Adapter for filter crucibles 3 Watch glas dish Ø 100 mm Ø*l = 19*170 mm 1 Graduated pipette 10 ml 1 Rubber spacers for 1 Quickfitt, complete 1 Feed pipe 1 Burette 25 ml filtering (screw thread adapter+seal 1 Pycnometer 25 ml 1 Weighing bottle, flat shape 1 Rubber sleeve for filter +srew-cap) 1 Vacuum tubing 30 ml crucible 1 Funnel holder (4 parts) øa*øi *l = 16*6*800 mm 1 Evaporating dish 109-0 (Ah0) Left lay Right lay: Desks 49-64 emty, 3* Pipette bulb, rubber, grey in left lay 1 Wire gauze square, ceramic center 1 Device block (from wood) 1 Support ring ø 80 mm 2 Test tube 75*12 mm 1 Burette holder 12 Test tube 75*10 mm 1 Clamp, universal 1 Nickel crucible 1 Crucible tong 1 Glas -
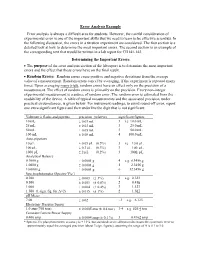
Error Analysis Example Error Analysis Is Always a Difficult Area for Students
Error Analysis Example Error analysis is always a difficult area for students. However, the careful consideration of experimental error is one of the important skills that we need to learn to be effective scientists. In the following discussion, the errors in a titration experiment are considered. The first section is a detailed look at how to determine the most important errors. The second section is an example of the corresponding text that would be written in a lab report for CH141-142. Determining the Important Errors The purpose of the error analysis section of the lab report is to determine the most important errors and the effect that those errors have on the final result. Random Errors: Random errors cause positive and negative deviations from the average value of a measurement. Random errors cancel by averaging, if the experiment is repeated many times. Upon averaging many trials, random errors have an effect only on the precision of a measurement. The effect of random errors is primarily on the precision. Every non-integer experimental measurement is a source of random error. The random error is estimated from the readability of the device. A table of typical measurements and the associated precision, under practical circumstances, is given below. For instrument readings, to avoid round-off error, report one extra significant figure and then underline the digit that is not significant. Volumetric flasks and pipettes precision (relative) significant figures 10 mL 0.03 mL 3 i.e. 10.0 mL 25 mL 0.03 mL 3 25.0 mL 50 mL 0.05 mL 3 50.0 mL 100 mL 0.08 mL 4 100.0 mL Auto-pipettors 10 L 0.05 L (0.5%) 3 i.e. -

Pipetting Lab Lab 3
Name:___________________________ Seat # ______ Do not write in pencil (use pen). Do not use Liquid Paper (draw one line through error). Date: ____/____/____ Periods_______________ Please staple all work. Pipetting Lab Lab 3 Purpose(s): The purpose of this lab is to learn how to pipette correctly so that accurate reagents can be made. Materials: 1. 1 Volumetric pipette 7. Test tube rack 2. 1 Ostwald pipette 8. 250 ml Beaker/flask of colored solution (one per student) 3. 1 Mohr pipette 9. 500 ml Beaker of water (one per two students) 4. 1 Serologic pipette 10. Kimwipe (paper towel) 5. 1 Pipette bulb 11. Work Mat ( Paper towel) 6. Test tubes (five per student) 12. Laboratory coat Procedure: Part 1 1. Select the volumetric pipette, and hold it in your dominant hand. 2. Place the pipette bulb in your non-dominant hand and squeeze it. 3. Attach the pipette bulb to the mouth of the pipette. 4. Place the tip of the pipette into the fluid and draw up fluid by releasing pressure on the pipette bulb. Make sure to draw up a little excess fluid pass the fill line, then place your index finger (still using your dominant hand) over the top of the pipette. Apply pressure so that the fluid does not leak from the pipette. 5. Wipe down the side of the pipette with a Kimwipe or paper towel. 6. Adjust the meniscus to the desired line of the pipette by gently releasing the pressure applied by your index finger until the liquid starts to flow out slowly. -

Poly-W109.01 Polyphenolics in Pulp Mill Wastewaters By
NCASI METHOD POLY W109.01 POLYPHENOLICS IN PULP MILL WASTEWATERS BY SPECTROPHOTOMETRY NCASI West Coast Regional Center November 2009 This page intentionally left blank. Acknowledgments This method was prepared by Ron Messmer, Senior Research Associate, and Diana Cook, Project Leader, at the NCASI West Coast Regional Center. Other assistance was provided by Jan Napack, Research Associate, also with the NCASI West Coast Regional Center. For more information about this method, contact: Diana Cook Project Leader NCASI West Coast Regional Center PO Box 458 Corvallis, OR 97339-0458 (541) 752-8801 [email protected] For information about NCASI publications, contact Publications Coordinator NCASI PO Box 13318 Research Triangle Park, NC 27709-3318 (919) 941-6400 [email protected] National Council for Air and Stream Improvement, Inc. (NCASI). 2009. Methods Manual. NCASI Method Poly W109.01: Polyphenolics in pulp mill wastewaters by spectrophotometry. Research Triangle Park, NC: National Council of the Paper Industry for Air and Stream Improvement, Inc. © 2009 by the National Council for Air and Stream Improvement, Inc. NCASI’s Mission To serve the forest products industry as a center of excellence for providing technical information and scientific research needed to achieve the industry’s environmental goals. i Disclaimer The mention of trade names or commercial products does not constitute endorsement or recommendation for use. This method is included in NCASI’s Methods Manual and is a modified Folin reagent method for tannin-lignin (polyphenols) determination in wastewater. It is included in the Methods Manual as a proposed method. Those electing to apply the method are strongly encouraged to conduct rigorous QA/QC or validation so that the quality of data generated can be evaluated. -

Rheologic Studies on Synovial Fluid
RHEOLOGIC STUDIES ON SYNOVIAL FLUID Milton G. Levine, David H. Kling J Clin Invest. 1956;35(12):1419-1427. https://doi.org/10.1172/JCI103399. Research Article Find the latest version: https://jci.me/103399/pdf RHEOLOGIC STUDIES ON SYNOVIAL FLUID By MILTON G. LEVINE AND DAVID H. KLING (From the Boyar-Kling Arthritis Clinic, Los Angeles, Calif.) (Submitted for publication March 28, 1956; accepted September 5, 1956) The relative viscosity of normal and pathologi- for most joint fluids; a 22 gauge is preferable for fluids cal synovial fluids fluctuates over a wide range, of low viscosity, and an 18 gauge for highly viscid fluids. and has been studied for a considerable period. The attachment of the needle to the pipette is made water-tight by a rubber sleeve, or by a cement seal, or The older literature is covered by Kling (1), and the delivery end of the pipette may be ground so that it the more recent by Ropes and Bauer (2) and by fits snugly into the hilt of the needle. Sundblad (3). Early investigators established, The viscometer is filled by suction above the zero by precipitation reactions with acetic acid, that mark, and the upper opening of the pipette is kept closed the high viscosity was due to a "mucin," assumed with a finger. The pipette is placed in a clamp holder and immersed in a constant temperature bath until the to be a combination of protein and carbohydrate. hilt of the hypodermic needle meets the water level. The In 1939, Meyer, Smyth, and Dawson (4) identi- measurement is started by releasing the finger, and the fied the polysaccharide as hyaluronic acid, and rate of flow is timed between two selected points on the found it to be responsible for the high viscosity. -

ORA Laboratory Manual Volume IV Section 1 06/30/2020 Title: Laboratory Orientation Page 1 of 35
OOD AND RUG DMINISTRATION Revision #: 02 F D A Document Number: OFFICE OF REGULATORY AFFAIRS Revision Date: IV-01 ORA Laboratory Manual Volume IV Section 1 06/30/2020 Title: Laboratory Orientation Page 1 of 35 Sections in This Document 1. Introduction............................................................................................................................... 2 2. FDA Laws and Regulations ......................................................................................................3 2.1. FD&C Act and Amendments ..........................................................................................3 2.2. Other Acts Enforced by FDA ..........................................................................................4 2.3. Code of Federal Regulations .........................................................................................7 2.4. Federal Register .............................................................................................................7 3. Analytical Methods ....................................................................................................................7 3.1. Official Compendia .........................................................................................................7 3.2. Specialized Manuals ......................................................................................................8 3.3. Other Method Sources ...................................................................................................9 3.4. Method Validation -

Volumetric Flask
Chemical Analysis Solution (Consumables) Nobel Laboratory Sdn. Bhd. Contents Beaker ............................................ pg. 01 Flask ............................................ pg. 02 Disk ............................................ pg. 05 Tube ............................................ pg. 07 Bottle ............................................ pg. 09 Funnel ............................................ pg. 11 Measure ............................................ pg. 13 Plastic Product ............................................ pg. 17 Rubber Product ............................................ pg. 27 Metalwares ............................................ pg. 31 Laboratory Accessories ............................................ pg. 34 Glassware Our glassware are made of high quality borosilicate glass, with competitive price and good quality. Beaker Low Type Beaker Description Specication Low Type Beaker 5-10000ml High Type Beaker Description Specication High Type Beaker 50-2000ml Conical Beaker Description Specication Conical Beaker 125-800ml Beaker with Handle Description Specication Beaker with Handle 100-1000ml 01 Flask Flat Bottom Flask Description Specication Flat Bottom Flask 5-20000ml Flat Bottom Flask (Ground Neck) 100-1000ml, Joint 14, 19, 24, 29, 40, 50 Round Bottom Flask Description Specication Round Bottom Flask 5-20000ml Round Bottom Flask (Ground Neck) 10-10000ml, Joint 14, 19, 24, 29, 40, 50 Kjeldahl Flask Description Specication Kjeldahl Flask 250-1000ml 02 Conical / Erlenmeyer Flask Description Specication