Uranium Recovery from Wet-Process Phosphoric Acid
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chemicals Used for Chemical Manufacturing Page 1 of 2
Chemicals used for Chemical Manufacturing Page 1 of 2 Acetic Acid (Glacial, 56%) Glycol Ether PMA Acetone Glycol Ether PNB Acrylic Acid Glycol Ether PNP Activated Carbon Glycol Ether TPM Adipic Acid Glycols Aloe Vera Grease Aluminum Stearate Gum Arabic Aluminum Sulfate Heat Transfer Fluids Amino Acid Heptane Ammonium Acetate Hexane Ammonium Bicarbonate Hydrazine Hydrate Ammonium Bifluoride Hydrochloric Acid (Muriatic) Ammonium Chloride Hydrogen Peroxide Ammonium Citrate Hydroquinone Ammonium Hydroxide Hydroxylamine Sulfate Ammonium Laureth Sulfate Ice Melter Ammonium Lauryl Sulfate Imidazole Ammonium Nitrate Isobutyl Acetate Ammonium Persulfate Isobutyl Alcohol Ammonium Silicofluoride Calcium Stearate Dipropylene Glycol Isopropanolamine Ammonium Sulfate Carboxymethylcellulose Disodium Phosphate Isopropyl Acetate Antifoams Caustic Potash D'Limonene Isopropyl Alcohol Antifreeze Caustic Soda (All Grades) Dodecylbenzene Sulfonic Acid Isopropyl Myristate Antimicrobials Caustic Soda (Beads, Prills) (DDBSA) Isopropyl Palmitate Antimony Oxide Cetyl Alcohol Dowfrost Itaconic Acid Aqua Ammonia Cetyl Palmitate Dowfrost HD Jojoba Oil Ascorbic Acid Chlorine, Granular Dowtherm SR-1 Keratin Barium Carbonate Chloroform Dowtherm 4000 Lactic Acid Barium Chloride Chromic Acid EDTA Lanolin Beeswax Citric Acid (Dry and Liquid) EDTA Plus Lauric Acid Bentonite Coal Epsom Salt Lauryl Alcohol Benzaldehyde Cocamide DEA Ethyl Acetate Lecithin Benzoic Acid Copper Nitrate Ethyl Alcohol (Denatured) Lime Benzyl Alcohol Copper Sulfate Ethylene Glycol Linoleic Acid Bicarbonate -

Inventory Stock Status by Item September 1 - 7, 2017 on Hand
10:14 AM MSU Chem Stores 09/07/17 Inventory Stock Status by Item September 1 - 7, 2017 On Hand Inventory 1-Propanol 1L (1-Propanol 1L) 2.00 1butanolt1L (tert-Butanol 1L Ea) 0.00 aceticacidgla (Acetic acid glacial 99.7% 2.5L Ea) 1.00 aceticanhyd (Acetic anhydride 500ml Ea) 0.00 aceton4Lchrom (Acetone chromAR for HPLC use) 0.00 acetone1L (Acetone HPLC 1L Ea) 0.00 acetone4L (Acetone HPLC 4L Ea) 1.00 acetonebulk (Acetone 345 lb/Drum (ACS) Bulk Lb) 720.63 Acetonit500ml (Acetonitrile 500ml ACS) 0.00 acetonitril4L (Acetonitrile HPLC 4L Ea 99.9%) 4.00 acetonitril4LHPLC99.9% (Acetonitril 4L HPLC 99.9% Gold Optima) -2.00 alconox (Alconox 4 Lb Carton Ea) 0.00 alcotabs (Alcotabs Bottle/100 Ea) 11.00 alumnitrate500 (Aluminum Nitrate 9 hydrate 500 g) 1.00 ammbicarb250 (Ammonium bicarbonate 250 gramEa) 0.00 ammcarbonaACS (Ammonium carbonate ACS 500g Ea) 1.00 ammchlori500g (Ammonium chloride 500g Ea) 0.00 ammfluoride (Ammonium fluoride 100g Ea) 0.00 ammhydroxide (Ammonium hydroxide 28-30% 2.5L Ea) 5.00 ammoniumnitrate2500 (Ammonium Nitrate Crystal 2500g) 8.00 ammoniumnitrate500 (Ammonium Nitrate Crystal 500g) 4.00 Ammoxal500g (Ammonium Oxalate 500G-Lab Grade) 0.00 amylalcmix8pt (Amyl alcohol (mixed isomers) 8pt (3.8L) Ea) 1.00 applicator (Applicators cotton-tipped Pk100) 77.00 aprondisp (Apron Tyvec 27x36" Ea) 1.00 ascorbicacid100g (L-ascorbic acid cry acs 100 grams) 0.00 aspiratorpoly (Aspirator poly Ea) 17.00 bags10x18 (Bags poly 10x18 Belart Ea) 90.00 bags12x20 (Bags poly 12x20 Belart Ea) 53.00 bagsbiohaz24x30 (Bags Biohazard 24"x30" 1.5mil per) 0.00 -

A Study on Physical Chemistry of Solid a Mmonium Materials for Nox Reduction of Diesel Engine Emissions
A Study on Physical Chemistry of Solid A mmonium Materials for NOx Reduction of Diesel Engine Emissions Cheon Seog (Steve) Yoon and Jong Kook Shin Hannam University, Daejeon, KOREA Hoyeol Lee and Hongsuk Kim Korea Institute of Machinery & Materials, Daejeon, KOREA 2014 DOE CLEERS Workshop University of Michigan, Dearborn, MI, USA 1 Table of Contents • Introduction of Solid SCR System • Ammonium Salts • Chemical Reactions, Decomposition Chemistry • Chemical Kinetic Parameters by TGA, DTA and DSC • Decomposition Rate from Hot Plate Test and Chemical Kinetic Parameters • Simple Reactor with Visible Window • Equilibrium Vapor Pressure Curve for Ammonium Carbonate • Acquisition of Re-solidified Materials from Ammonium Carbonate • Analytical Study of Re-solidified Materials from Ammonium Carbonate by XRD, FT-IR, and EA • Concluding Remarks • Acknowledgement • Reference 2 Solid SCR System • NOx purification technology by using NH3, which is generated from solid ammonium. • Ammonium carbonate, (NH4)2CO3 , is solid at room temperature, and it decomposes into NH3, H2O & CO2 above temperature of 60℃. 3 Material Properties of Ammonium Salts Solid urea Ammonium carbonate Ammonium cabarmate Molecular formula (NH2)2CO (NH4)2CO3 NH2COONH4 Molecular weight 60.07 96.09 78.07 3 Density, g/cm 1.33 1.5 1.6 Mols NH3 per Mol 2 2 2 Mols NH3 per kg 33.3 20.8 25.6 Decomposition temp., ℃ 140 58 60 NH2CONH2↔ NH3+HNCO Reaction mechanism (NH4)2CO3↔2NH3+CO2+H2O NH4COONH2 ↔ 2NH3 + CO2 HNCO +H2O ↔ NH3 + CO2 Cost cheap cheap moderate * HNCO: Isocyanic Acid [ref] G. Fulks, -

Urea: a Low Cost Nitrogen Fertilizer with Special Management Requirements 1
Urea: A Low Cost Nitrogen Fertilizer with Special Management Requirements 1 may loose from 50% to 90% of its N as ammonia if not Urea: A Low Cost protected within a few hours of application. Nitrogen Fertilizer with Conserving Urea-N Special Management If the urea-to-NH4 reaction takes place in the soil the Requirements nitrogen will be captured as exchangeable ammonium on the soil exchange complex and little if any loss of ammonia gas to the air will occur. Therefore, the key to conserving urea fertilizer nitrogen is to put the urea into the soil and not merely on the soil. Soil incorporation of urea can be done several ways. Since urea is completely water soluble, when applied to the soil surface it can be moved down with irrigation water or rainfall, if one or the other occurs immediately after fertilization. Also, urea can be broadcast and plowed down immediately. And urea can be injected or banded into the soil. Urea (46-0-0) usually has the lowest cost per pound Soil banding or injection is usually not feasible with an of nitrogen compared to other single-element nitrogen established crop such as pasture. Under these conditions fertilizers. However, urea undergoes unique chemical the nitrogen fertilizer of choice would be either ammonium transformations when field applied and severe losses in nitrate, ammonium sulfate, or one of the ammoniated efficiency may result if special management practices are phosphates (for example 11-52-0). not followed. The purpose of this fact sheet is to briefly describe urea transformations and to suggest how urea-N Urea is applied alone or in combination with other may be conserved with proper management in the field. -

Disposal of Solid Chemicals in the Normal Trash
Disposal of Solid Chemicals in the Normal Trash Many solid chemicals can be safety discarded into the normal trash, provided they are in containers that are not broken or cracked and have tightly fitting caps. These chemicals are considered acceptable for ordinary disposal because they display none of the properties of hazardous waste, are of low acute toxicity, and have not been identified as having any chronic toxic effects as summarized in the National Institute of Occupational Safety and Health (NIOSH) “Registry of Toxic Effects of Chemical Substances”. Examples of chemicals acceptable for disposal as regular trash are listed below. To dispose of these chemicals, place the containers in a box lined with a plastic bag, tape the top of the box shut, write “Normal Trash” on the box and then place the box next to the lab trash container. Only solid forms of these chemicals can be disposed in this manner. Any questions about these chemicals or other chemicals that may be disposed of in the normal trash should be directed to the Hazardous Materials Technician (610) 330-5225. Chemicals Generally Acceptable for Disposal as Regular Trash Acacia powder, gum Detergent (most) Methyl salicylate Sodium carbonate arabic Cation exchange resins Methylene blue Sodium chloride Acid, Ascorbic Chromatographic Methyl stearate Sodium citrate Acid, Benzoic absorbents Nutrient agar Sodium dodecyl sulfate Acid, Boric Crystal violet Octacosane (SDS) Acid, Casamind Dextrin Parafin Sodium formate Acid, Citric Dextrose Pepsin Sodium iodide Acid, Lactic Diatomaceous -

Solid Chemicals for the Normal Trash
SOLID CHEMICALS FOR THE NORMAL TRASH Solid chemicals acceptable for disposal as regular trash are listed below: Acacia powder, gum arabic Calcium silicate Manganese sulfate Acid, Ascorbic Calcium sulfate Methyl red Acid, Benzoic Detergent (most) Methyl salicylate Acid, Boric Cation exchange resins Methylene blue Acid, Casamind Crystal violet Methyl stearate Acid, Citric Dextrin Nutrient agar Acid, Lactic Dextrose Octacosane Acid, Oleic Diatomaceous earth Parafin Acid, Phthalic Docosanoic acid Pepsin Acid, Salicycle Drierite (calcium sulfate, anhydrous) Peptone Acid, Silicic Ferric oxide Petroleum jelly Acid, Stearic Ferric phosphate Polyethylene, solid Acid, Succinic Ferric pyrophosphate Polystryene Acid, Tartaric Ferric sulfate Potassium acetate Acrylamide gels Ferrous ammonium sulfate Potassium bicarbonate Agar(s) Galactose Potassium bromide Albumen Geletin Potassium carbonate Alumina Gum arabic Potassium chloride Aluminum oxide Gum guaiac Potassium citrate Amino acids, naturally occurring Hexadecanol, 1- Potassium ferricyanide Ammonium bicarbonate Kaolin Potassium iodide Ammonium phosphate Lactose Potassium phosphate Ammonium sulfate Lanolin Potassium sodium tartrate Ammonium sulfamate Lauric acid Potassium sulfate Base, blood agar Lauryl sulfate Potassium sulfite Beef extract Lithium carbonate Potassium sulfocyanate Behenic acid Lithium chloride Pumice Bentonite Lithium sulfate Salts, naturally occurring Brain heart infusion Litmus Sand Bromphenol blue Magnesium carbonate Silica Broth, nutrient Magnesium chloride Silica gel, unused -
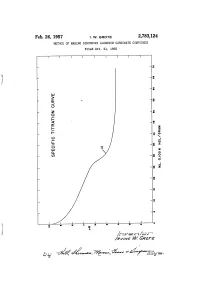
Feb. 26, 1957 I 1
Feb. 26, 1957 I 1. w. GROTE 2,783,124 METHOD OF MAKING DIHYDROXY ‘ALUMINUM CARBONATE COMPOUNDS‘ ' Filed Oct. 21, 19755 ‘ Ohn.O.inOnonn00»ON6'0d0-O-n0 . 2,783,124 States Patent 'icc Patented Feb. 26, 1957 2 present invention are preferably the aluminum alkoxides, such as aluminum isopropylate (tri-isopropoxide), but 2,783,124 other aluminum tri-alkoxides may be used in which the alkoxide groups are C2-C4 groups. These aluminum al METHOD OF MAKING DEHYDROXY ALUMINUM coholates are hydrolyzable in an alkaline aqueous medi~ CARBQNATE CDMPOUNDS um to form aluminum hydroxide or other aluminum lrvine W. Grote, Chattanooga, Tenn., assignor to The group containing at least two hydroxyl radicals, the other Chattanooga Medicine Company, Chattanooga, Tenn, valency of the aluminum being satis?ed by, possibly, an a corporation of Tennessee alkoxide radical. 10 The reaction involved in the case of the aluminum Application October 21, 1955, Serial No. 542,023 alcoholates is represented by the following equation: 11 Claims. (Cl. 23-14) (1) ‘ (ZHzO) . A1(OR)3 + M11003 ---> (H0)2A1o COzM+3ROH wherein R is a C2-C4 alkyl group and M is a monovalent This invention relates to a method of preparing di 15 radical of the class consisting of alkali metal and am hydroxy aluminum carbonate compounds, and more par monium radicals. It is probable, as before stated, that ticularly to a method of making dihydroxy aluminum the reaction involves an intermediate formation of di sodium, potassium, or ammonium carbonates. These are hydroxy aluminum alcoholate. This reaction is prefera true compounds having the generic structural formula bly carried out in the presence of equi-molar proportions hereinafter given. -

List of Lists
United States Office of Solid Waste EPA 550-B-10-001 Environmental Protection and Emergency Response May 2010 Agency www.epa.gov/emergencies LIST OF LISTS Consolidated List of Chemicals Subject to the Emergency Planning and Community Right- To-Know Act (EPCRA), Comprehensive Environmental Response, Compensation and Liability Act (CERCLA) and Section 112(r) of the Clean Air Act • EPCRA Section 302 Extremely Hazardous Substances • CERCLA Hazardous Substances • EPCRA Section 313 Toxic Chemicals • CAA 112(r) Regulated Chemicals For Accidental Release Prevention Office of Emergency Management This page intentionally left blank. TABLE OF CONTENTS Page Introduction................................................................................................................................................ i List of Lists – Conslidated List of Chemicals (by CAS #) Subject to the Emergency Planning and Community Right-to-Know Act (EPCRA), Comprehensive Environmental Response, Compensation and Liability Act (CERCLA) and Section 112(r) of the Clean Air Act ................................................. 1 Appendix A: Alphabetical Listing of Consolidated List ..................................................................... A-1 Appendix B: Radionuclides Listed Under CERCLA .......................................................................... B-1 Appendix C: RCRA Waste Streams and Unlisted Hazardous Wastes................................................ C-1 This page intentionally left blank. LIST OF LISTS Consolidated List of Chemicals -

Carbonate, and Ammonium Oxalate to Assess the Availability of Molybdenum in Mine Spoils and Soils'
COMPARISON OF AMMONIUM BICARBONATE-DTPA, AMMONIUM CARBONATE,AND AMMONIUM OXALATE TO ASSESS THE AVAILABElTY OF MOLYBDENUM IN MINE SPOILS AND SOILS L. Wag, KJ. Reddy and LC. Munn Journal Article 1994 WWRC-94-03 In Comun. Soil Sci. Plant Anal. L. Wag, KJ. Reddy and L.C. MUM Wyoming Water Resources Center University of Wyoming Laramie, Wyoming t COMMUN. SOIL SCI. PLANT ANAL., 25(5&6), 523-536 (1994) J COMPARISON OF AMMONIUM BICARBONATE-DTPA, AMMON- IUM CARBONATE, AND AMMONIUM OXALATE TO ASSESS THE AVAILABILITY OF MOLYBDENUM IN MINE SPOILS AND SOILS' L. Wangl, K, J. Reddy, and L. C. Munn' Water Resources Center, University of Wyoming,Laramie, WY 82071 pBSTRACT: A variety of extractants has been used to assess the availability of molybdenum (Mo) in soils. Most of the extractants have been studied from a deficiency aspect rather than for soils with Mo toxicity, and none of them have been used to extract available Mo from mine spoils. The purpose of this study was to examine the potential of different chemical extractants for assessing the availability of Mo in mine spoils and soils. One mine spoil and three soils were treated with sodium molybdate and then subjected to wetting and drying cycles for two months. These spoiVsoils were extracted with ammonium bicarbonate DTPA (AB-DTPA), ammonium carbonate, and ammonium oxalate solution for available Mo. Crested wheatgrass (Agropyroncristatum) and alfalfa (Medicagosativa) were grown in the spoil/soils in a greenhouse to determine plant uptake of Mo. Additionally, four mine spoils and six soils were extracted and analyzed for available Mo as mentioned above. -

Kaolin Sodium Bicarbonate
March 31, 2020 Ms. Michelle Arsenault National Organic Standards Board USDA-AMS-NOP 1400 Independence Ave. SW Room 2648-S, Mail Stop 0268 Washington, DC 20250-0268 Docket ID # AMS-NOP-19-0095 Re. HS: Sunset 605 These comments to the National Organic Standards Board (NOSB) on its Spring 2020 agenda are submitted on behalf of Beyond Pesticides. Founded in 1981 as a national, grassroots, membership organization that represents community-based organizations and a range of people seeking to bridge the interests of consumers, farmers and farmworkers, Beyond Pesticides advances improved protections from pesticides and alternative pest management strategies that reduce or eliminate a reliance on pesticides. Our membership and network span the 50 states and the world. Kaolin Reference: 205.605(a) Kaolin is a fine clay, consisting primarily of hydrous aluminum silicate. Because of the small particle size, it has a high surface-to-volume ratio, making it a highly absorptive material. Although the TAP review identifies it as an anticaking agent and a processing aid that is not present in the final product, there is no annotation to limit its use. Kaolin is also produced in nano-sized particles.1 Kaolin should be annotated to specify allowed uses and prohibit the use of nano-kaolin. Sodium bicarbonate Reference: 205.605(a) Baking soda is the kind of material that was envisioned as populating the National List— a nontoxic natural material used in home kitchens as a leavening agent. 1 https://www.researchgate.net/publication/297841906_The_properties_of_Nano-kaolin_mixed_with_kaolin. Waxes –wood rosin Reference: 205.605(a) Waxes—nonsynthetic (Wood resin). -

Ajax Finechem Product Catalogue 3510 585 6
ACE AJAX FINECHEM PRODUCT CATALOGUE 3510 ACETONE, HPLC GRADE, BURDICK & JACKSON, CAT. AH010 Assay ……………………………………………………….. 99.9% min. Maximum limit of impurities (%) Water ……………………………………………………….. 0.5 Residue ……………………………………………………… 3mg/L Max. UV. Absorbance: λ(nm) 330 340 350 375 400 Absorbance 1.000 0.080 0.010 0.005 0.005 Pack Size: 4L 585 ACETONE, SPECTROSOL Description: clear liquid; characteristic odour. For U.V. spectroscopy. Assay (by GLC) ……………………………………………….. 99.5% min. Colour (APHA) …………………………………............ 10 max. Density (@ 25°C ) ……………………………………… 0.7857g/mL max. U.V absorbance λ(nm) 330 340 350 400 Max abs. 1.00 0.1 0.02 0.01 Maximum limit of impurities (%) R.A.E. ……………………………………………………….. 0.001 Sol. in H2O ………………………………………………… To pass test Titratable acid …………………………………………… 0.03 mmol H Titratable base ………………………………………….. 0.06 mmol OH Aldehyde (as HCHO) ………………………………….. 0.002 CH3OH ………………………………………………………. 0.05 Propan-2-ol ……………………………………………….. 0.05 Subs. red. KMnO4 (as O) …………………………….. 0.0005 H2O ………………………………………………………….. 0.5 Conforms to ACS Pack Size: 500mL 6 ACETONE, UNIVAR Description: clear liquid with a characteristic odour. Assay( by GLC) ………………………………………….. 99.5% min. Colour (APHA) …………………………………………… 10 max. Density (@ 25oC) ……………………………….......... 0.7857g/mL max. Maximum limit of impurities (%) R.A.E. ……………………………………………………….. 0.001 Cd …………………………………………………. 0.000005 Sol. in H2O …………………………………………………. To pass test Pb ………………………………………………….. 0.000005 Titratable acid ……………………………………………. 0.03 mmol H Ca………………………………………………….. 0.00005 Titratable base …………………………………………… 0.06 mmol OH Zn ………………………………………………….. 0.00005 Aldehyde (as HCHO) …………………………………… 0.002 Na …………………………………………………. 0.00005 Methanol, Propan-2-ol (each) ………………………. 0.05 K …………………………………………………….. 0.00005 Fe …………………………………………………………….. 0.00002 Cr…………………………………………………… 0.000002 Subs. red. KMnO4`……………………………………….. To pass test Co ………………………………………………….. 0.000002 H2O …………………………………………………………… 0.5 Cu ………………………………………………….. 0.000002 Al ……………………………………………………………… 0.00001 Mn …………………………………………………. -

Development of an Analytical Method for Distinguishing
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by TopSCHOLAR Western Kentucky University TopSCHOLAR® Masters Theses & Specialist Projects Graduate School 1-1-2004 Development of an Analytical Method for Distinguishing Ammonium Bicarbonate from the Products of an Aqueous Ammonia Co2 Scrubber and the Characterization of Ammonium Bicarbonate Lingyu Meng Western Kentucky University Follow this and additional works at: http://digitalcommons.wku.edu/theses Part of the Environmental Chemistry Commons Recommended Citation Meng, Lingyu, "Development of an Analytical Method for Distinguishing Ammonium Bicarbonate from the Products of an Aqueous Ammonia Co2 Scrubber and the Characterization of Ammonium Bicarbonate" (2004). Masters Theses & Specialist Projects. Paper 243. http://digitalcommons.wku.edu/theses/243 This Thesis is brought to you for free and open access by TopSCHOLAR®. It has been accepted for inclusion in Masters Theses & Specialist Projects by an authorized administrator of TopSCHOLAR®. For more information, please contact [email protected]. DEVELOPMENT OF AN ANALYTICAL METHOD FOR DISTINGUISHING AMMONIUM BICARBONATE FROM THE PRODUCTS OF AN AQUEOUS AMMONIA CO2 SCRUBBER AND THE CHARACTERIZATION OF AMMONIUM BICARBONATE A Thesis Presented to The Faculty of the Department of Chemistry Western Kentucky University Bowling Green, Kentucky In Partial Fulfillment of the Requirements for the Degree Master of Science by Lingyu Meng December, 2004 DEVELOPMENT OF AN ANALYTICAL METHOD FOR DISTINGUISHING AMMONIUM BICARBONATE FROM THE PRODUCTS OF AN AQUEOUS AMMONIA CO2 SCRUBBER AND THE CHARACTERIZATION OF AMMONIUM BICARBONATE Date Recommended 12/15/2004 Dr. Wei-Ping Pan, Director of Thesis Dr. Stuart Burris Dr. M. Thandi Buthelezi Elmer Gray, Dean of the Graduate College, (December 16, 2004) ACKNOWLEDGEMENT I would like to express my deep appreciation to Dr.